DEPARTMENT OF ENVIRONMENT, GREAT LAKES, AND ENERGYENVIRONMENTAL QUALITY AIR QUALITY DIVISION AIR POLLUTION CONTROL Filed with the secretary of state on These rules become effective immediately after filing with the secretary of state unless adopted under section 33, 44, or 45a(9) of the administrative procedures act of 1969, 1969 PA 306, MCL 24.233, 24.244, or 24.245a. Rules adopted under these sections become effective 7 days after filing with the secretary of state.
(By authority conferred on the director of the department of environment, Great Lakes, and energyenvironmental quality by sections 5503 and 5512 of the natural resource and environmental protection act, 1994 PA 451, MCL 324.5503 and 324.5512, and Executive Reorganization Order Nos. 1995-168, 2009-31, 2011-1, 2019-1, MCL 324.99903, 324.99919, 324.99921, and 324.99923) R 336.2001, R 336.2003, R 336.2004, R 336.2011, R 336.2012, R 336.2014, R 336.2033, R 336.2040, and R 336.2041 of the Michigan Administrative Code are amended, as follows: PART 10. INTERMITTENT TESTING AND SAMPLING R 336.2001 Performance tests by owner.Rule 1001. (1) The department may require the owner or operator ofanya source of air contaminant to conduct acceptable performance tests, at the owner's or operator's expense, in accordance with R 336.2003 under any1of the following conditions:
(a)Prior toBefore issuance of a permit to operate.
(b) The source is determined to be in violation of R 336.1301 and the potential emissions exceed 100 tons per year.(c) The owner or operator of the source has not submitted an acceptable performance test, in accordance with R 336.2003, that demonstrates that the sourceis in compliancecomplies with either the department's rulesandorwiththe conditions specified in the permit to install, or both.
(d) The source of air contaminant is located in an area designated as nonattainment for 1 or more air pollutants, and more than 12 months have expired since the date of the last performance test for such designated nonattainment pollutants.(e) The source of air contaminant has potential emissions in excess of 100 tons per year, is located in an area designated as attainment for 1 or more air pollutants, and more than 36 months have expired since the date of the last performance test forsuchthe designated attainment pollutants.
(f) After completion of a compliance program.(2) Performance tests required by subrule (1) of this ruleshallmust be conducted within 60 days following receipt of written notification from the department, unless otherwise authorized by the department.
(3) Not less than 30 days beforeFora performance test, as required by subrule (1) of this rule, the owner or operator, or their authorized agent, shall do both of the following:
(a)sSubmit a site-specific test plannot less than 30 days before a performance testfor approvalofby the department. The planwillmust include a test program summary, test schedule, and the quality assurance measures to be applied.
(4)(b)Not less than 7 days before performance tests are conducted, the owner of a source of air contaminant, or his or her authorized agent, shall nNotify the department, in writing, of the time and place of the performance tests and who shall conduct them as provided in the site-specific test plan required under subdivision (a) of this subrule. A representative of the department shall have the opportunity to witness these tests.
(54) Results of performance testsshallmust be submitted to the department in the format prescribed by the applicable reference test method within 60 days after the last date of the test.
R 336.2003 Performance test criteria.Rule 1003. (1) Performance testsshallmust be conducted and data reduced according to the reference test methods listed in R 336.2004, unless the department does any of the following:
(a) Specifies or approves, in specific cases, the use of a reference test method with minor changes in procedures or equipment. (b) Approves the use of an equivalent method. (c) Specifies or approves the use of an alternative method if an applicable reference test method does not exist for a specific air contaminant or source of air contaminant.(2) Unless otherwise approved by the department, aAperformance testshallmust consist of a minimum of 3 separate samples of a specific air contaminant conducted within a 36-hour period that starts once the probe enters the stack. Any data measured within the 36-hour period must be recorded and provided to the department.unless otherwise authorized by the department.Each of the 3 separate samplesshallmust be obtained while the source is operating at a similar production level, as described under subrule (3) of this rule. For the purpose of determining compliance with an applicable emission limit, rule, or permit condition, the arithmetic mean of results of the 3 samplesshallmust apply. If a sample is accidentally lost or conditions occur in which 1 of the 3 samples must be discontinued because of forced shutdown, failure of an irreplaceable portion of the sampling train, extreme meteorological conditions, or other circumstances beyond the owner's or operator's control, then compliance may, upon the approval of the department, be determined using the arithmetic mean of the results of 2 samples.
(3) All performance testsshallmust be conducted while the source of air contaminant is operating at maximum routine operating conditions, or undersuchother conditions, within the capacity of the equipment, as may be requested by the department. Other conditions may include source operating periods of startup, shutdown, orsuchother operations, excluding malfunction, specific to certain sources. Routine operating conditionsshallmust also include those specified within a permit to install or a permit to operate. The owner or operator shall make available to the departmentsuchthe recordsasthat may be necessary to determine the conditions of source operation that occurred during the period of time of the performance test.
(4) Foranysources thatisare subject to an emission limitation calculated to 50% excess air, the multipoint, integrated sampling procedure of method 3shallmust be used for gas analysis. For all other sources that require a determination of the molecular weight of the exhaust,anyan optional sampling procedure of method 3 may be used. Alternatives or modifications to procedures are subject to the approval of the department.
(5) For reference test methods5B, which is described in R 336.2011,andreference test method 5C, which is described in R 336.2012, and reference test method 5E, which is described in R 336.1014, the minimum volume per sampleshallmust be 30 cubic feet of dry gas corrected to standard conditions,(70 degrees Fahrenheit, 29.92 in.chesHgmercury). Minimum sample timeshallmust be 60 minutes, which may be continuous or a combination of shorter sampling periods for sources that operate in a cyclic manner. Smaller sampling times or sample volumes, when necessitated by process variables or other factors, may be approved by the department.
R 336.2004Appendix A; rReference test methods; adoption of federal reference test methods.
Rule 1004. (1) Thefollowingfederalreferencetest methods,described in the provisions of 40C.F.R.CFR part 60, appendix A (2007), adopted by reference in R 336.1902, arethe referencetest methods for performance tests required pursuant to the provisions of this part and include, but are not limited to, the following:
(a) Method 1 - Sample and velocity traverse for stationary sources. (b) Method 1A - Sample and velocity traverses for stationary sources with small stacks or ducts. (c) Method 2 - Determination of stack gas velocity and volumetric flow rate (type-S pitot tube). (d) Method 2A - Direct measurement of gas volume through pipes and small ducts. (e) Method 2C - Determination of stack gas velocity and volumetric flow rate in small stacks and ducts (standard pitot tube). (f) Method 2D - Measurement of gas volumetric flow rates in small pipes and ducts. (g) Method 3 - Gas analysis for the determination of dry molecular weight. (h) Method 4 - Determination of moisture content in stack gases.(i) Method 5 - Determination of particulate matter emissions from stationary sources.
(j) Method 6 - Determination of sulfur dioxide emissions from stationary sources. (k) Method 7 - Determination of nitrogen oxide emissions from stationary sources. (l) Method 8 - Determination of sulfuric acid mist and sulfur dioxide emissions from stationary sources. (m) Method 9 - Visual determination of the opacity of emissions from stationary sources. (n) Method 10 - Determination of carbon monoxide emissions from stationary sources. (o) Method 10B - Determination of carbon monoxide emissions from stationary sources. (p) Method 18 - Measurement of gaseous organic compound emissions by gas chromatography. (q) Method 21 - Determination of volatile organic compound leaks.(r) Method 24 - Determination of volatile matter content, water content, density, volume solids, and weight solids of surface coatings.
(s) Method 24A - Determination of volatile matter content and density of printing inks and related coatings. (t) Method 25 - Determination of total gaseous nonmethane organic emissions as carbon. (u) Method 25A - Determination of total gaseous organic concentration using a flame ionization analyzer. (v) Method 27 - Determination of vapor tightness of gasoline delivery tank using pressure-vacuum test. (w) Method 29 - Determination of metals emissions from stationary sources. (x) Method 30A - Determination of total vapor phase mercury emissions from stationary sources (instrumental analyzer procedure). (y) Method 30B - Determination of total vapor phase mercury emissions from coal-fired combustion sources using carbon sorbent traps.(2) The federal test methods in the following provisions, adopted by reference in R 336.1902, are test methods for performance tests required pursuant to the provisions of this part:
(a) 40 CFR part 51, appendix M. (b) 40 CFR part 61, appendix B. (c) 40 CFR part 63, appendix A.The reference test methods listed in subrule (1) of this rule are adopted by reference in this rule. Copies of the test methods may be inspected at the Lansing office of the air quality division of the department of environmental quality. A copy of title 40 of the Code of Federal Regulations, part 60, appendix A, may be obtained from the Department of Environmental Quality, Air Quality Division, P.0. Box 30260, Lansing, Michigan 48909 7760, at a cost at the time of adoption of these rules of $67.00; from the Superintendent of Documents, United States Government Printing Office, P.O. Box 979050, St. Louis, Missouri 63197-9000, at a cost at the time of adoption of these rules of $57.00; or on the United States government printing office internet web site at http://www.gpoaccess.gov.
(3) All alternatives that are subject to the approval of the administrator in the adopted federal reference methods are subject to the approval of the department.(4) Determinations of compliance with visible emission standards for stationary sourcesshallmust be conducted as specified in 40 CFR part 60, appendix A,referencetest method 9 orotheranother alternative method approved by the department, with the following exceptions:
(a) Visible emissions from a scarfing operation at a steel manufacturing facilityshallmust be determined as specified in reference test method 9A, which is described in R 336.2030.
(b) Visible emissions from a coke oven pushing operation and fugitive coke oven visible emissionsshallmust be determined as specified in reference test method 9B, which is described in R 336.2031.
(c) Visible emissions, fugitive and nonfugitive, from basic oxygen furnace operations, hot metal transfer operations, and hot metal desulfurization operationsshallmust be determined as specified in reference method 9C, which is described in R 336.2032.
(5) Determinations of particulate emission rates for stationary sourcesshallmust be conducted as specified in 1 or more of the following reference test methods:
(a) Reference test method 5B, which is described in R 336.2011. (b) Reference test method 5C, which is described in R 336.2012. (c) Reference test method 5D, which is described in R 336.2013. (d) Reference test method 5E, which is described in R 336.2014.(e) "Standard Methods for the Examination of Water and Wastewater," (14th23rd edition),section 208C,as described and modified in R 336.2033.
(6) Determinations of total gaseous nonmethane organic emissions as carbon, using the alternate version of federalreferencetest method 25 under 40 CFR part 60, appendix A, incorporating the Byron analysis,shallmust be conducted as specified in R 336.2006.
R 336.2011 Reference test method 5B.
Rule 1011. Reference test method 5B, in-stack filtration method, reads as follows:
(a) The principle, applicability, and performance test criteria are as follows:
(i) Principle. Particulate matter is withdrawn isokinetically from the source and collected on solid filtering media maintained at stack temperature. The particulate matter mass is determined gravimetrically after removal of uncombined water.
(ii) Applicability. This method is applicable for the determination of particulate emissions from stationary sources as identified in table 31 of R 336.1331. The method is also applicable when specifically provided for in the department’s rules, orders, a permit to install, or a permit to operate.
(iii) Performance test criteria as follows:
(A) A performance test mustshallconsist meet the
requirements under R 336.2003(2). of a minimum of 3 separate samples of a
specific air contaminant conducted within a 36-hour period, unless otherwise authorized
by the department. Each of the 3 separate samples shall be obtained while the
source is operating at a similar production level. For the purpose of
determining compliance with an applicable emission limit, rule, or permit
condition, the arithmetic mean of results of the 3 samples shall apply. If a
sample is accidentally lost or conditions occur in which 1 of the 3 samples
must be discontinued because of forced shutdown, failure of an irreplaceable portion
of the sampling train, extreme meteorological conditions, or other
circumstances beyond the owner's or operator's control, compliance may, upon
the approval of the department, be determined using the arithmetic mean of the
results of 2 samples.
(B) For any sources that are is subject
to an emission limitation calculated to 50% excess air, the multipoint,
integrated sampling procedure of R 336.2004(1)(c) shall must be used for gas
analysis. For all other sources that require a determination of the molecular
weight of the exhaust, any an optional sampling procedure of R
336.2004(1)(c) may be used. Alternatives or modifications to procedures are
subject to the approval of the department.
(C) The minimum volume per sample shall
must be 30 cubic feet
of dry gas corrected to standard conditions, (70 degrees
Fahrenheit, and 29.92 inches
mercury). Minimum sample time shall must be 60 minutes,
which may be continuous or a combination of shorter sampling periods for
sources that operate in a cyclic manner. Smaller sampling times or sample
volumes, if necessitated by process variables or other factors, may be approved
by the department.
(D) For any a source whose
emission control device alters the moisture content of the exhaust gas, a
moisture determination shall must be performed in a location
upstream from the emission control device and in accordance with R
336.2004(1)(d) or an alternative method approved by the department.
(b) The following provisions apply to apparatus:
(i) Sampling train. A schematic of the
sampling train used in this method is shown in figure 102 under R 336.2021.
Construction details for many, but not all, of the train components are given
in APTD-0581,
adopted by reference in R 336.1902. (See subdivision (g)(ii) of this
rule.) For changes from the APTD-0581 document and for allowable
modifications to figure 102, the user shall consult with the department. The
operating and maintenance procedures for many, but not all, of the sampling
train are described in APTD-0576, adopted by reference in R 336.1902. (See
subdivision (g)(iii) of this rule.) Since correct usage is important in
obtaining valid results, all users shall read APTD-0576 and adopt the
applicable operating and maintenance procedures outlined in it, unless
otherwise specified herein. The sampling train shall must consist of the
following components:
(A) Probe nozzle. Stainless steel (316)
or glass with sharp, tapered leading edge. The angle of taper shall must
be less than 30 degrees and the taper shall must be on the
outside to preserve a constant internal diameter. The probe nozzle shall
must be of the button-hook design, unless otherwise specified by the
department. If made of stainless steel, the nozzle shall must be
constructed from seamless tubing. Other materials of construction may be used,
subject to the approval of the department. A range of nozzle sizes suitable for
isokinetic sampling shall must be available, for example, 0.32 to
1.27 centimeters, (1/8 to 1/2 inch,).
or larger if higher volume sampling trains are used inside diameter (ID)
nozzles in increments of 0.16 centimeters, (1/16 inches).
Each nozzle shall must be calibrated according to the procedures
outlined in subdivision (e) of this rule.
(B) Probe liner. Interior surface may
be constructed of stainless steel, (no specific grade),
glass, teflonTeflon, or such other
material that maintains proper flow at the stack conditions experienced.
(C) Pitot tube. Type S, as described
in section 2.1 of method 2, or other device approved by the department. The
pitot tube shall must be attached to the probe, as shown in
figure 102 under R 336.2021, to allow constant monitoring of the stack
gas velocity. The impact, (high pressure,) opening plane of
the pitot tube shall must be even with or above the nozzle entry
plane, (see
method 2, figure 2-6b,) during sampling. The
type S pitot tube assembly shall must have a known coefficient,
determined as outlined in section 4 of method 2.
(D) Differential pressure gauge. Two incline Incline
manometer or equivalent devices (2) as described in section 2.2 of
method 2. One manometer shall must be used for velocity head (p)
readings and the other shall must be used for orifice differential
pressure readings.
(E) Filter holders. Two separate
filter holders in series or 1 filter holder with separate filter supports and
seals for 2 filters. One filter holder with 2 filters held in contact with each
other is not acceptable. Materials of construction may be stainless steel (316),
glass, teflonTeflon, or other
material approved by the department.
(F) Filter heating system. Auxiliary
heating of the filter media is not acceptable. For saturated stack gases, the
operator may opt to use filters that do not blind when wet and that do not
require heating, (see
subdivision (c)(i)(A) of this rule).
(G) Condenser. The following system shall
must be used to determine the stack gas moisture content: Three
impingers connected in series with leak-free ground glass fittings or any similar
leak-free noncontaminating fittings. All impingers shall must be
of the Greenburg-Smith design and shall must be modified by
replacing the tip with a 1.3 centimeters, (1/2 inch,)
ID inside diameter glass tube extending to about 1.3 centimeters,
(1/2 inch,) from the bottom of the flask. Modifications,
such as using flexible connections between the impingers or using materials
other than glass, are permitted allowed, subject to the approval
of the department. The first impinger shall must contain a known
quantity of water, (as described in subdivision (d)(i)(C)
of this rule.); the The second impinger shall
must be empty;, and the third shall must
contain a known weight of silica gel or equivalent desiccant. Alternatively, any
a system that cools the sample gas stream and allows measurement of
the water condensed and moisture leaving the condenser, each to within 1 milliliter
or 1 gram, may be used, subject to the approval of the department. In
any case, the means for measuring the moisture leaving the condenser shall
must be by passing the sample gas stream through a tared silica gel, or
equivalent desiccant, trap with exit gases kept below 20 degrees Centigrade,
(68 degrees Fahrenheit,) and determining the weight
gain. If a determination of the particulate matter collected in the impingers
is required by the department’s rules, a permit to install, or a permit to
operate, then the impinger system described above shall must be
used without modification. Contact the department as to the sample recovery and
analysis of the impinger contents.
(H) Metering system. Vacuum gauge,
leak-free pump, thermometers capable of measuring temperature to within 3
degrees Centigrade, (5.4
degrees Fahrenheit), dry-gas meter capable of measuring volume to within
2%, and related equipment as shown in figure 102 under R 336.2021. Other
metering systems capable of maintaining sampling rates within 10% of isokinetic
and capable of determining sample volumes to within 2% may be used, subject to
the approval of the department. When the metering system is used in conjunction
with a pitot tube, the system shall must enable checks of
isokinetic rates. Sampling trains utilizing metering systems designed for
higher flow rates than those described in APTD-0581 or APTD-0576, both adopted by
reference in R 336.1902, may be used if the specifications of this method are
met.
(I) Barometer. Mercury, aneroid, or
other barometer capable of measuring atmospheric pressure to within 2.5 millimeters
Hgmercury, (0.1 inch Hg mercury).
In many cases, the barometric reading may be obtained from a nearby national
weather service station. In this case, the station value, which is the absolute
barometric pressure, shall must be requested and an adjustment
for elevation differences between the weather station and sampling point shall
must be applied at a rate of minus 2.5 millimeters Hgmercury,
(0.1 inch Hg mercury,) per 30 Mmeters,
(100 feet.), elevation increase or vice versa for
elevation decrease.
(J) Gas density determination
equipment. Temperature sensor and pressure gauge, as described in sections
2.3 and 2.4 of method 2, and gas analyzer, if necessary, as described in
method 3. The temperature sensor shall must, preferably, be
permanently attached to the pitot tube or sampling probe in a fixed
configuration such so that the tip of the sensor extends beyond the leading
edge of the probe sheath and does not touch any metal. Alternatively, the
sensor may be attached just before use in the field. If the temperature sensor
is attached in the field, then the sensor shall must be placed in an
interference-free arrangement with respect to the type S pitot tube openings, (see
method 2, figure 2-76 Velocity Traverse Data). As a second
alternative, if a difference of not more than 1% in the average velocity
measurement is to be introduced, then the temperature gauge need not be
attached to the probe or pitot tube. This alternative is subject to the
approval of the department. “Construction Details of Isokinetic Source Sampling
Equipment,” APTD-0581, April 1971, (PB203-060-LL), and
“Maintenance, Calibration, and Operation of Isokinetic Source Sampling
Equipment,” APTD-0576, March 1972, (PB209-022-LL), are adopted
by reference in R
336.1902.
this rule. Copies of these documents may be inspected at the Lansing office of
the air quality division of the department of environmental quality. Copies of
APTD-0581 and APTD-0576 may be obtained from the Department of Environmental
Quality, Air Quality Division, P.O. Box 30260, Lansing, Michigan 48909-7760, or
from the National Technical Information Service, U.S. Department of Commerce, 5285
Port Royal Road, Springfield, Virginia 22161, at a cost at the time of adoption
of these rules of $28.50 each.
(ii) Sample recovery. The following items are required:
(A) Probe-liner and probe-nozzle
brushes. Nylon bristle brushes with stainless steel wire handles. The probe
brush shall
must
have extensions, at least as long as the probe, made of stainless steel, nylon,
teflonTeflon, or similarly
inert material. The brushes shall must be properly sized and shaped to
brush out the probe liner and nozzle.
(B) Wash bottles 2. Glass wash bottles are recommended; the tester may use polyethylene wash bottles, but the acetone should not be stored in polyethylene bottles for longer than 1 month.
(C) Glass sample storage containers. Chemically
resistant, borosilicate glass bottles for acetone washes, 500 milliliters
or 1000 milliliters. Screw cap liners shall must
either be rubber-backed teflonTeflon or shall must
be constructed so as to be leak-free and resistant to chemical attack by
acetone. Narrow-mouth glass bottles are less prone to leakage. Alternatively,
polyethylene bottles may be used.
(D) Filter containers. Glass, polyethylene, or aluminum tube containers, unless otherwise specified by the department.
(E) Graduated cylinder or balance. To
measure condensed water to within 1 milliliter or 1 gram.,
graduated cylinders shall must have subdivisions of not more than
2 milliliters. Most laboratory balances are capable of weighing
to the nearest 0.5 gram or less. Any of these balances may be used here
and in paragraph (iii)(D) of this subdivision.
(F) Plastic storage containers. Airtight containers to store silica gel.
(G) Funnel and rubber policeman. To
aid in the transfer of silica gel to container;, but not necessary if
silica gel is weighed in the field.
(H) Funnel. Glass or polyethylene, to aid in sample recovery.
(iii) Analysis. The following equipment is required for analysis:
(A) Glass weighing dishes.
(B) Desiccator.
(C) Analytical balance. To measure to
within 0.1 milligrams.
(D) Balance. To measure to within 0.5
milligrams.
(E) Beakers. 250 milliliters.
(F) Hygrometer. To measure the relative humidity of the laboratory environment.
(G) Temperature gauge. To measure the temperature of the laboratory environment.
(c) The following provisions shall must apply to
reagents:
(i) Sampling. The reagents used in sampling are as follows:
(A) Filters. Two in-stack filters may
be any combination of alundum ceramic thimble filters, type RA-98, or glass
fiber filters, type A without organic binder. The size of such the
filters shall must allow proper sampling rates to maintain iso-kinetics
using the nozzle sizes specified in subdivision (b)(i)(A) of this rule. Alternatively,
other types of filters may be used, subject to the approval of the department.
(B) Silica gel. Indicating type, 6 to
16 mesh. If previously used, dry at 175 degrees Centigrade, (350 degrees
Fahrenheit,) for 2 hours. New
silica gel may be used as received. Alternatively, other types of desiccants
that are equivalent or better may be used, subject to the approval of the
department.
(C) Water. When analysis of the
material caught in the impingers is required, distilled water shall must be used. Run
blanks before field use to eliminate a high blank of test samples.
(D) Crushed ice.
(E) Stopcock grease.
Acetone-insoluble, heat-stable silicone grease. This is not necessary if
screw-on connectors with teflon Teflon sleeves, or equivalent, are used. Alternatively,
other types of stopcock grease may be used, subject to the approval of the
department.
(ii) Sample recovery. Washing solvent.
Either acetone or distilled water may be used for sample recovery. If acetone
is used for washing solvent, then reagent grade, less than 0.001% residue, in glass
bottles is required. Acetone from metal containers generally has a high residue
blank and shall
must
not be used. If suppliers transfer acetone to glass bottles from metal
containers, then acetone blanks shall must be run before
field use and only acetone with low blank values, (less than
0.001%,) shall must be used. In no case shall must a blank value of
more than 0.001% of the weight of acetone used be subtracted from the sample
weight. If distilled water is used for washing solvent, use distilled water
with less than 0.001% residue. Run blanks before field use to eliminate a high
blank on test samples.
(iii) Analysis. Two reagents are required for the analysis:
(A) Solvent. Same as paragraph (ii) of this subdivision for quantitative transfer.
(B) Desiccant. Anhydrous calcium sulfate, indicating type. Alternatively, other types of desiccants may be used, subject to the approval of the department.
(d) The following provisions shall must apply to
procedure:
(i) Sampling. The complexity of this
method is such that, in order to obtain reliable results, testers shall be
trained and experienced with the test procedures. Sampling shall must comply with the
following provisions:
(A) Pretest preparation. provisions are as
follows:
(I). All the
components shall must be maintained and calibrated according to
the applicable procedures described in APTD-0576, adopted by
reference in R 336.1902, unless otherwise specified in this rule.
(II) Weigh several 200 to 300 gram portions of silica gel in airtight containers to the nearest 0.5 gram. Record the total weight of the silica gel plus container on each container. As an alternative, the silica gel need not be preweighed, but may be weighed directly in its impinger or sampling holder just before train assembly.
(III) Check filters
visually against light for irregularities, flaws, pinhole leaks, or cracks. Label
filters of the proper size on the back side using numbering machine ink. As an
alternative, label the shipping containers, as described under (subdivision
(b)(ii)(D) of this rule), and keep the filters in these containers
at all times, except during sampling and weighing.
(IV) Dry the filters
in an oven at 105 degrees Centigrade, (220 degrees Fahrenheit,) for a minimum of
2 hours, cool for at least 1 hour in a desiccator containing anhydrous calcium
sulfate, and individually weigh and record their weights to the nearest 0.1 milligram. During the
weighing, the filter shall must not be exposed to the laboratory
atmosphere for a period of more than 2 minutes and a relative humidity above
50%.
(V) Procedures, other than those specified, that account for relative humidity effects may be used, subject to the approval of the department.
(B) Preliminary determinations.
provisions
are as follows:
(I) Select the sampling site and the minimum number by the department.
(II) Determine the
stack pressure, temperature, and the range of velocity heads using method 2;. it It is recommended
that a leak check of the pitot lines, (see method 2, section 3.1A) be performed.
(III) Determine the moisture content using approximation method 4, or its alternatives, for the purpose of making isokinetic sampling rate settings.
(IV) Determine the
stack gas dry molecular weight, as described in method 2, section 3.6;
if integrated method 3 sampling is used for molecular weight determination, the
integrated bag sample shall must be taken simultaneously with, and
for the same total length of time as, the particulate sample run.
(V) Select a nozzle
size based on the range of velocity heads so that it is not necessary to change
the nozzle size to maintain isokinetic sampling rates. During the run, do not
change the nozzle size. Ensure that the proper differential pressure gauge is
chosen for the range of velocity heads encountered, (see section
2.2 of method 2).
(VI) Select a suitable probe liner and probe length so that all traverse points may be sampled. For large stacks, sampling from opposite sides of the stack may reduce the length of probes.
(VII) Select a total
sampling time greater than or equal to the minimum total sampling time
specified in the department’s rules so that the sampling time per point is not
less than 5 minutes, unless approved by the department, or some greater time
interval as specified by the department, and so that the sample volume taken,
corrected to standard conditions, exceeds the required minimum total gas sample
volume. The latter is based on an approximate average sampling rate. The number
of minutes sampled at each point may be an integer or an integer plus 1/2
minute to avoid timekeeping errors. In some circumstances, such as in batch
cycles, it may be necessary to sample for shorter times at the traverse points
and to obtain smaller gas sample volumes. In these cases, the department’s
approval shall
must
first be obtained.
(C) Preparation of collection train. provisions are as
follows:
(I) During preparation and assembly of the sampling train, keep all openings where contamination can occur covered until just before assembly or until sampling is about to begin.
(II) Place 100 milliliters of water in the
first impinger, leave the second impinger empty, and transfer approximately 200
to 300 grams of preweighed
silica gel from its container to the third impinger. More silica gel may be
used, but care shall must be taken to ensure that it is not
entrained and carried out from the impinger during sampling. Place the
container in a clean place for later use in the sample recovery. Alternatively,
the weight of the silica gel plus impinger may be determined to the nearest 0.5
gram and recorded.
(III) Using tweezers or
clean disposable surgical gloves, place a labeled, (identified,) and weighed
filter in each filter holder. Be sure that the filter is properly centered and
the gasket properly placed so as to prevent the sample gas stream from
circumventing the filter.
(IV) Install the
selected nozzle using a Viton A 0-ring when stack temperatures are less than
260 degrees Centigrade, (500
degrees Fahrenheit,) and an asbestos
string gasket when temperatures are higher. See APTD-0576, adopted by
reference in R 336.1902, for requirements. Other connecting systems using
either 310 stainless steel or teflon Teflon ferrules may be
used to form a leak-free direct mechanical connection.
(V) Mark the probe with heat-resistant tape or by some other method to denote the proper distance into the stack or duct for each sampling point.
(VI) Set up the train as in figure 102 under R 336.2021.
(VII) If necessary, use
a very light coat of silicone grease on all ground glass joints. Grease only
the outer portion, (see
APTD-0576,) to avoid the
possibility of contamination by the silicone grease.
(VIII) Place crushed ice
around the impingers.
(D) Leak check procedures:
(I1) Pretest leak
check. A pretest leak check is strongly recommended, but not required, to prevent
invalid sampling and wasted time. If the tester opts to conduct the pretest
leak check, the following procedure shall must be used: Perform
the leak check on the entire system, including filter housings and nozzle, by
plugging the nozzle and pulling a 380 millimeter Hgmercury,
(15 inch Hgmercury,) vacuum. Alternatively,
a lower vacuum may be used if it is not exceeded during the test. Leakage rates
in excess of 4% of the average sampling rate or 0.00057 m³/mincubic
meters per minute, (0.02 cfmcubic feet per minute),
whichever is less, are unacceptable. The following leak check instructions for
the sampling train described in APTD-0576 and APTD-0581, adopted by
reference in R 336.1902, may be helpful. Start the pump with the bypass
valve fully open and the coarse adjust valve completely closed. Partially open
the coarse adjust valve and slowly close the bypass valve until the desired
vacuum is reached. Do not reverse the direction of the bypass valve;,
as this will cause water to back up into the flexible sample tube and the
probe. If the desired vacuum is exceeded, either leak-check at this higher
vacuum or end the leak check and start over. When the leak check is completed,
first slowly remove the plug from the inlet to the nozzle and immediately turn
off the vacuum pump. This prevents the water in the first impinger from
being forced backward into the sample tube and prevents silica gel from being
entrained backward into the second impinger.
(II2) Leak checks
during sample run. If, during the sampling run, a component, (such as a
filter assembly or impinger,) change becomes necessary, a leak check shall must be conducted
immediately before the change is made. The leak check shall must be done according
to the procedure outlined in paragraph (i)(D)(1I) of this
subdivision, except that it shall must be done at a vacuum equal to or
greater than the maximum value recorded up to that point in the test. If the
leakage rate is not more than 0.00057 m³/mincubic meters per
minute,
(0.02 cfmcubic feet per minute,) or 4% of the
average sampling rate, whichever is less, then the results are acceptable and
no correction need be applied to the total volume of dry gas metered. If a
higher leakage rate is obtained, then the tester shall either record the
leakage rate and plan to correct the sample volume, as shown in subdivision
(f)(iii) of this rule, or shall void the sampling run. Immediately after
component changes, leak checks may be performed. If leak checks are done, then
the procedure outlined in paragraph (i)(D)(1I) of this
subdivision shall must be used.
(III3) Post-test leak
check. A leak check is required at the conclusion of each sampling run. The
leak check shall must be performed in accordance with the
procedures in paragraph (i)(D)(1I) of this subdivision, except that it shall must be conducted at a
vacuum equal to or greater than the maximum value reached during the sampling
run. If the leakage rate is not more than 0.00057 m³/mincubic meters per
minute,
(0.02 cfmcubic feet per minute,) or 4% of the
average sampling rate, whichever is less, then the results are acceptable and
no correction need be applied to the total volume of dry gas metered. If a
higher leakage rate is obtained, then the tester shall either record the
leakage rate and correct the sample volume, as shown in subdivision (f)(iii) of
this rule, or shall void the sampling run.
(E) Particulate train operation. During
the sampling run, maintain an isokinetic sampling rate that is within 10% of
true isokinetic, unless otherwise specified by the department. For each run,
record the data required on a data sheet such as the data sheet in figure 104
under R 336.2021. Record the initial dry-gas meter reading. Record the
dry-gas meter readings at the beginning and end of each sampling time
increment, when changes in flow rates are made, before and after each leak
check, and when sampling is halted. Take other readings required by figure 104
under R 336.2021 at least once at each sample point during each time
increment, and take additional readings when significant changes, (20%
variation in velocity head readings,) necessitate additional
adjustments in flow rate. Level and zero the manometer. Because the manometer
level and zero may drift due to vibrations and temperature changes, make
periodic checks during the traverse. Clean the portholes before the test run to
minimize the chance of sampling deposited material. To begin sampling, remove
the nozzle cap and verify that the pitot tube and probe are properly
positioned. Position the nozzle at the first traverse point with the tip
pointing directly into the gas stream. Immediately start the pump and adjust
the flow to isokinetic conditions. Nomographs that aid in the rapid adjustment
of the isokinetic sampling rate without excessive computations are available. These
nomographs are designed for use when the type S pitot tube coefficient is 0.85
±0.02 and the stack gas equivalent density, (dry molecular weight,)
is equal to 29 ±4. APTD-0576, adopted by reference in R 336.1902, details
the procedure for using the nomographs. If Cp and Md are outside the above
stated ranges, do not use the nomographs unless appropriate steps, (see
subdivision (g)(iv) of this rule,) are taken to compensate for
the deviations. When the stack is under significant negative pressure, (height
of impinger stem), take care to pull low-flow when inserting the probe
into the stack to prevent water from backing into the sample tubing and to avoid
pulsation through the filter and possible loss of materials. When the probe is
in position, block off the openings around the probe and porthole to prevent
unrepresentative dilution of the gas stream. Traverse the stack cross section,
as required by method 1 or as specified by the department, being careful not to
bump the probe nozzle into the stack walls when sampling near the walls or when
removing or inserting the probe through the portholes; this minimizes the
chance of extracting deposited material. During the test run, add more ice and,
if necessary, salt to maintain a temperature of less than 20 degrees Centigrade,
(68 degrees Fahrenheit,) at the condenser/silica gel
outlet. Also, periodically check the level and zero of the manometer. If the
pressure drop across the filter becomes too high and makes isokinetic sampling
difficult to maintain, the filter may be replaced in the midst of a sample run.
It is recommended that another complete filter assembly be used rather than
attempting to change the filter itself. Before a new filter assembly is
installed, conduct a leak check, (as described under paragraph
(i)(D)(2II) of this subdivision). The total particulate
weight shall must include the summation of all filter assembly
catches. A single train shall must be used for the entire sample
run, except in cases where simultaneous sampling is required in 2 or more
separate ducts, at 2 or more different locations within the same duct, or where
equipment failure necessitates a change of trains. In all other situations, the
use of 2 or more trains shall must be subject to the approval of
the department. When 2 or more trains are used, separate analyses of the
front-half and, if applicable, impinger catches from each train shall
must be performed, unless identical nozzle sizes were used on all trains. If
identical nozzle sizes were used, the front-half catches from the individual
trains may be combined, as may the impinger catches, and 1 analysis of
front-half catch and 1 analysis of impinger catch may be performed. Consult
with the department for details concerning the calculation of results when 2 or
more trains are used. At the end of the sample run, turn off the coarse adjust
valve, remove the probe and nozzle from the stack, turn off the pump, record
the final dry-gas meter reading, and conduct a post-test leak check, as
outlined in paragraph (i)(D)(3III) of this subdivision. Leak-check
the pitot lines as described in method 2., section 3.1; the The
lines shall must pass this leak check to validate the velocity
head data.
(F) Calculation of percent
isokinetic. Calculate percent isokinetic, (see subdivision (f) of this rule,) to determine if
the run was valid or if another test run should be made. If there was
difficulty in maintaining isokinetic rates due to source conditions, consult
with the department for possible variance on the isokinetic rates.
(ii) Sample recovery. Proper cleanup
procedure begins as soon as the probe is removed from the stack at the end of
the sampling period. Allow the probe to cool. When the probe can be safely
handled, wipe off all external particulate matter near the tip of the probe
nozzle and place a cap over it to prevent losing or gaining particulate matter.
Do not cap off the probe tip tightly while the sampling train is cooling down
as this creates a vacuum in the filter holder and draws water from the
impingers into the sample tube. Before moving the sampling train to the cleanup
site, make sure all condensed water in the probe and flexible sample lines are
drained into the first impinger. Disconnect all sample lines and remove the
nozzle-filter set assembly from the probe. Cap all openings to prevent
contamination or accidental loss of sample. Remove all excess particulate from
the exterior of the nozzle-filter assembly to prevent contamination during
disassembly. Transfer the nozzle-filter set assembly and impinger set to the
cleanup area. The cleanup area shall must be clean and
protected from the wind so that the chances of contaminating or losing the
sample are minimized. Save a portion of the solvent used for cleanup as a
blank. Take 200 milliliters of this solvent
directly from the wash bottle being used and place it in a glass sample
container labeled "solvent blank". Inspect the train before and
during disassembly and note any abnormal conditions. Treat the samples in the
following manner: Container Nnumbersos. 1, 1A. Carefully
remove the filters from the filter holders and place in their identified
containers. Use a pair of tweezers or clean disposable surgical gloves, or
both, to handle the filters. Carefully transfer to the container any particulate
matter or filter fibers, or both, that adhere to the filter holder gasket by
using a dry nylon bristle brush or sharp-edged blade, or both. Seal the
containers. Container numberNo. 2. Taking care to
see that particulate on the outside of the nozzle and filter holders does not
get into the sample, the testoer shall carefully remove the nozzle and
clean the inside surface by rinsing with solvent from a wash bottle and
brushing with a nylon bristle brush. Brush until the solvent rinse shows no
visible particles and then make a final rinse of the inside surface with
solvent. After ensuring that all joints have been cleaned of all extraneous
material, the testoer shall quantitatively remove particulate from the
filter holders by rubbing the surfaces with a nylon bristle brush and rinsing
with solvent. Rinse each surface 3 times, or more if needed, to remove visible
particulate. Make a final rinse of the brush and filter holder set. After all solvent
washings and particulate matter have been collected in the sample container,
tighten the lid on the sample container so that solvent will not leak out when
it is shipped to the laboratory. Mark the height of the fluid level to
determine if leakage occurred during transport. Label the container to clearly
identify its contents. Container numberNo. 3. Note the color
of the indicating silica gel to determine if it has been completely spent and
make a notation of its condition. Transfer the silica gel from the third
impinger to its original container and seal. A funnel may make it easier to
pour the silica gel without spilling it. A rubber policeman may be used as an
aid in removing the silica gel from the impinger. It is not necessary to remove
the small amount of dust particles that adhere to the impinger wall and are
difficult to remove. Since the gain in weight will be used for moisture
calculations, do not use any water or other liquids to transfer the silica gel.
If a balance is available in the field, follow the procedure for container numberNo. 3 in paragraph
(iii) of this subdivision. Impinger water. Treat the impingers in the following
manner: Make a notation of any color or film in the liquid catch. Measure the
liquid that is in the first 2 impingers to within ±1 milliliter by using a
graduated cylinder or by weighing it to within ±1.0 g gram by using a
balance if one is available. Record the volume or weight of liquid present. This
information is required to calculate the moisture content of the effluent gas. Discard
the liquid after measuring and recording the volume or weight, unless analysis
of the impinger catch is required, (see subdivision
(b)(i)(G) of this rule). If a different type of condenser is used,
measure the amount of moisture condensed either volumetrically or
gravimetrically. If possible, containers shall must be shipped in a
manner that keeps them upright at all times.
(iii) Analysis. Record the data
required on a sheet such as the sheet in figure 106 under R 336.2021. Handle
each sample container in the following manner: Container numbersNos.
1, 1A. Analyze and report each filter separately. Transfer the filter and any
loose particulate from the sample container to a tared-glass weighing dish. Dry
the filter in an oven at 105 degrees Centigrade, (220 degrees
Fahrenheit,) for a minimum of 2 hours, cool for at least 1 hour
in a desiccator containing anhydrous calcium sulfate, and weigh and
record its weight to the nearest 0.1 milligram. During the
weighing the filter shall must not be exposed to the laboratory
atmosphere for a period greater than 2 minutes or a relative humidity above
50%. Procedures, other than those specified, that account for relative humidity
effects may be used, subject to the approval of the department. The method used
for drying and weighing of filters shall must be consistent
before and after the test. Container No.number 2. Note the level
of liquid in the container and confirm on the analysis sheet if leakage
occurred during transport. If a noticeable amount of leakage has occurred, then
either void the sample or use methods, subject to the approval of the
department, to correct the final results. Measure the liquid in this container
either volumetrically to ±1 millileter or gravimetrically to ±1.0
gram. Transfer the contents to a tared 250-millileter
beaker and evaporate to dryness either at ambient temperature and pressure for
acetone or at 95 degrees Centigrade, (203 degrees Fahrenheit,)
in an oven for distilled water. Then subject the sample to 250 degrees
Centigrade, (482 degrees Fahrenheit,) in an oven for 2 to
3 hours. Desiccate 24 hours and weigh to a constant weight. Report the results
to the nearest 0.1 milligram. Container No.number
3. Weigh the spent silica gel, or silica gel plus impinger, to the nearest 0.5
gram using a balance. This step may be conducted in the field. "Solvent
blank" container. Measure solvent in this container either volumetrically
or gravimetrically. Transfer the contents to a tared 250-milliliters
beaker and evaporate to dryness either at ambient temperature and pressure for
acetone or at 95 degrees Centigrade, (203 degrees Fahrenheit,)
in an oven for distilled water. Then subject the sample to 250 degrees
Centigrade, (482 degrees Fahrenheit,) in an oven for 2 to
3 hours. Desiccate for 24 hours and weigh to a constant weight. Report the
results to the nearest 0.1 mg milligram. If acetone is used, the
contents of Container No.number 2, as well as the acetone blank
container, may be evaporated at temperatures higher than ambient. If
evaporation is done at an elevated temperature, then the temperature shall
must be closely supervised, and the contents of the beaker shall
must be swirled occasionally to maintain an even temperature. Use extreme
care, as acetone is highly flammable and has a low flash point.
(e) Calibration. Maintain a laboratory
log of all calibrations. Calibrations shall must comply with the
following provisions:
(i) Probe nozzle. Probe nozzles shall must be calibrated
before their initial use in the field. Using a micrometer, measure the inside
diameter of the nozzle to the nearest 0.025 millimeter, (0.001 in.ch). Make 3 separate
measurements using different diameters each time and obtain the average of the
measurements. The difference between the high and low numbers shall must not exceed 0.1 millimeter, (0.004 in.ch). When nozzles
become nicked, dented, or corroded, they shall the nozzles must be reshaped,
sharpened, and recalibrated before use. Each nozzle shall must be permanently
and uniquely identified.
(ii) Pitot tube. The type S pitot tube
assembly shall
must
be calibrated according to the procedures in section 4 of method 2.
(iii) Metering system. Before its
initial use in the field, the metering system shall must be calibrated
according to the procedure in APTD-0576, adopted by reference in R 336.1902. Instead of
physically adjusting the dry-gas meter dial readings to correspond to the
wet-test meter readings, calibration factors may be used to mathematically
correct the gas meter dial readings to the proper values. Before calibrating
the metering system, a leak check may be conducted. For metering systems having
diaphragm or rotary pumps, the normal leak check procedure will not detect
leakages within the pump. For these cases, the following leak check procedure
may be used: Make a 10-minute calibration run at 0.00057 m³/mincubic meters per
minute,
(0.02 cfmcubic feet per minute.); at At the end of the
run, take the difference of the measured wet-test meter and dry-gas meter
volumes; and divide the
difference by 10 to get the leak rate. The leak rate shall must not exceed
0.00057 m³/mincubic
meters per minute
(0.02 cfmcubic
feet per minute).
After each field use, the calibration of the metering system shall must be checked by
performing 3 calibration runs at a single, intermediate orifice setting, based
on the previous field test, with the vacuum set at the maximum value reached
during the test series. To adjust the vacuum, insert a valve between the
wet-test meter and the inlet of the metering system. Calculate the average
value of the calibration factor. If the calibration has changed by more than
5%, then recalibrate the meter over the full range of orifice settings, as
outlined in APTD?-0576. Alternatively, a spirometer may be substituted
for a wet-test meter in the above calibration procedures. Alternative
procedures, such as using the orifice meter coefficients, may be used, subject
to the approval of the department. If the dry-gas meter coefficient values
obtained before and after a test series differ by more than 5%, then the test
series shall
must
be performed using whichever meter coefficient value, (before or
after,) gives the lower
value of total sample volume.
(iv) Temperature gauges. Use the
procedure in section 4.3 of method 2 to calibrate in-stack temperature
gauges. Dial thermometers, such as those used for the dry-gas meter and
condenser outlet, shall must be calibrated against
mercury-in-glass thermometers.
(v) Leak check of metering system
shown in figure 102 under R 336.2021. That portion of the sampling train
from the pump to the orifice meter shall must be leak-checked
before initial use and after each shipment. Leakage after the pump will result
in less volume being recorded than is actually sampled. The following procedure
is suggested, also (see
figure 107 under R 336.2021): Close the main valve on the meter
box. Insert a 1-hole rubber stopper with rubber tubing attached into the
orifice exhaust pipe. Disconnect and vent the low side of the orifice
manometer. Close off the low side orifice tap. Pressurize the system to 13 to
18 centimeters, (5 to 7 in.ches), water column by
blowing into the rubber tubing. Pinch off the tubing and observe the manometer
for 1 minute. A loss of pressure on the manometer indicates a leak in the meter
box;. Leaks Leaks, if present, shall must be corrected.
(vi) Barometer. Calibrate against a mercury barometer.
(f) Calculations. When carrying out
calculations, retain at least 1 extra decimal figure beyond that of the
acquired data. Round off figures after the final calculation. Other forms of
the equations may be used if they the other forms of the equations give equivalent
results. The following provisions apply to calculations:
An = Cross-sectional area of nozzle, meters² (or the equivalent feet.²).
A = Cross-sectional area of stack or flue at the point of sampling, feet².
B ws = Water vapor in the gas stream, proportion by volume, expressed as a fraction.
B wi = Percent water vapor in gas entering source particulate control device determined by method 4.
B wo = Percent water vapor in gas exiting source particulate control device.
Ca = Wash blank residue concentration, milligrams per gram.
Cs = Concentration of particulate matter in stack gas, pounds per 1,000 pounds of actual stack gas.
C sD = Concentration of particulate matter in stack gas, moisture excluded, pounds per 1000 pounds of dry stack gas.
Cs50 = Concentration of particulate matter corrected to 50% excess air, pounds per 1000 pounds of stack gas.
Cs50D = Concentration of particulate matter corrected to 50% excess air, excluding any water addition from a collector, pounds per 1000 pounds of stack gas.
E = Mass emission rate of particulate, lbpounds/hour.
F50 = Concentration conversion factor to 50% excess air with no moisture alterations in exhaust.
F50D = Concentration conversion factor to 50% excess air, excluding any moisture added to exhaust gas by pollution collection system.
FD = Concentration conversion factor to dry basis, excluding any water in the stack gas.
I = Percent of isokinetic sampling.
L a = Maximum acceptable leakage rate for either a pretest leak check or for a leak check following a component change; equal to 0.00057 meters³/minute (0.02 cubic feet per minute) or 4% of the average sampling rate, whichever is less.
Li = Individual leakage rate observed during the leak check conducted
before the "ith" component change (i = 1, 2, 3 . . . . n), meters³/minute (cubic feet per minute).
Lp = Leakage rate observed during the post-test leak check, meters³/minute (cubic feet per minute).
Md = Molecular weight of dry stack gas, gram/gram mole (lbpound/lbpound-mole), calculated
by
method 3, equation 3-12, using data from integrated
method 3.
mn = Total amount of particulate matter collected, milligram.
Mw = Molecular weight of water, 18.0 gram/gram-mole (18.0 lbpound/lbpound-mole).
ma = Mass of residue of solvent after evaporation, milligram.
mg = Total weight of gas samples through nozzle, lbpound.
P bar = Barometric pressure at the sampling site, millimeter mercury Hg (inches Hgmercury).
Ps = Absolute stack gas pressure.
Pstd = Standard absolute pressure, 760 millimeters Hgmercury (29.92 inches Hgmercury).
R = Ideal gas constant, 0.06236 mm /°K-g-mole millimeters of
mercury-cubic meters per kelvin-gram-mole, (21.85 in.Hg-ft.³/R?lb-mole inches of
mercury-cubic feet per Rankine-pound-mole).
T m = Absolute average dry-gas meter temperature, see figure 104 under R 336.2021, °Kelvin, (°Rankine).
Ts = Absolute average stack gas temperature, see figure 104 under R 336.2021, °Kelvin, (°Rankine).
Tstd = Standard absolute temperature, 294.I°Kelvin, (530°Rankine).
V a = Volume of solvent blank, millileters.
V aw = Volume of solvent used in wash, millileters.
V lc = Total volume of liquid collected in impingers and silica gel (see figure 106 under R 336.2021), millileters.
Vm = Volume of gas sample as measured by the dry-gas meter, deci-centimeter, (deci-cubic-foot).
V m(std) = Volume of gas sample measured by the dry-gas meter, corrected to standard conditions, deci-standard cubic meter, (deci-standard cubic foot).
V w(std) = Volume of water vapor in the gas sample, corrected to standard conditions, standard cubic meter, (standard cubic foot).
V s = Stack gas velocity, calculated by method 2, equation
2-9, using data obtained from method 5, meters/second (feet/second).
Wa = Weight of residue in solvent wash, milligram.
Y = Dry-gas meter calibration factor.
ΔH = Average pressure differential across the orifice meter (see figure
104 under R 336.2021), millimeter water H20 (inches waterH20).
%02 = Percent oxygen in stack gas by volume (dry basis).
%N2 = Percent nitrogen in stack gas by volume (dry basis).
p a = Density of solvent, milligrams/millileter.
p s(std) = Density of all sampled gas at standard conditions, lbpounds/feet.³
pw = Density of water, 0.9982 grams/millileter (0.002201 lbpounds/millileter).
θ = Total sample time, minute.
θ1 = Sample time, interval, from the beginning of a run until the
first component change, minute.
θi = Sampling time interval, between 2 successive component changes, beginning with the interval between the first and second changes, minute.
θp = Sampling time interval, from the final (nth) component change until the end of the sampling run, minute.
13.6 = Specific gravity of mercury.
60 = Seconds/minute.
100 = Conversion to percent.
386.9 = Cubic feet per lbpound-mole of ideal gas at standard
conditions.
453.6 = Conversion of pounds to grams.
3600 = Conversion of hours to seconds.
1000 = Conversion of 1000 lbpound units to lbpound units.
(ii) Average the dry-gas meter
temperature and average the orifice pressure drop. See data sheet, (figure 5-2
104
under R 336.2021).
(iii) Dry gas volume. Correct the
sample volume measured by the dry-gas meter to standard conditions, (21.11
degrees Centigrade, 760 millimeters Hgmercury or 70 degrees
Fahrenheit, 29.92 in.ches Hgmercury,) by using equation
5-1.
Eequation 5-1:
![]()
Where:
K1 = 0.3869 °K/mm Hg for metric units.
= 17.71 °R/in. Hg for English units.
Equation 5-1 may be used as written.
However, if the leakage rate observed during any of the mandatory leak checks, (for
example, the post-test leak check or leak checks conducted before component
changes,) exceeds La,
equation 5-1
must
shall be modified as follows: in the following manner:
(A) Case I. No component changes made during sampling run. In this case, replace Vm in equation 5-1 with the following expression:
![]()
(B) Case II. One or more component changes made during the sampling run. In this case, replace Vm in equation 5-1 by the following expression:
![]()
and substitute only for those leakage rates (Li or Lp) that exceed La.
(iv) Volume of water vapor.
Eequation 5-2
![]()
Where:
K2 = 0.001338 m³/ml for metric units.
= 0.04733 ft.3/ml for English units.
(v) Moisture content.
Eequation 5-3
![]()
In saturated or water droplet-laden gas
streams, 2 calculations of the moisture content of the stack gas shall must
be made: 1 from the impinger analysis, (equation 5-3), and a
second from the assumption of saturated conditions. The lower of the 2 values
of Bws shall must be considered correct. The
procedure for determining the moisture content based upon on the assumption
of saturated conditions as described in is given in the note of section
1.2 of 40 CFR part 60 appendix A method 4. For the purpose of this
method, the average stack gas temperature from figure 104 under R 336.2021
may be used to make the determination, if the accuracy of the in-stack
temperature sensor is ±1 degree Centigrade, (2 degrees Fahrenheit).
(vi) Solvent blank concentration.
Eequation 5-4
![]()
(vii) Solvent wash blank.
Eequation 5-5
![]()
(viii) Total
particulate weight. Determine the total particulate catch from the sum of the
weights obtained from containers 1, 1A, and 2 less the wash solvent blank, (see
figure 106 under R 336.2021). Refer to subdivision (d)(i)(E) of
this rule to assist in the calculation of results involving 2 or more pairs of
filters or 2 or more sampling trains.
(ix) Sampled
gas density. Determine the density of the gas sampled from the stack, at
standard conditions in
pounds per cubic foot, (lb/ft.³).
Eequation 5-6
![]()
(x) Total
weight of gas sampled, (lbs).
Eequation 5-7
![]()
(xi) Particulate
concentration, (lbs/1000
lbs).
Eequation 5-8
![]()
(xii) Excess air and moisture correction factors:
(A) Correction
factor to 50% excess air for those sources with or without any a particulate
collector where no increase in moisture content of the exhaust gas occurs after
the process and before the point of sampling.
Eequation 5-9
![]()
(B) Correction factor to 50% excess
air for those sources with a wet collection device, (scrubber,) that increases
the moisture content of the exhaust gas after the process and before the point
of sampling.
Eequation 5-10
![]()
(C) Correction factor to convert the actual concentration, Cs, to dry conditions.
Eequation 5-11
![]()
(xiii) Converted particulate concentrations, where applicable under the department’s rules or permit.
Eequation 5-12
![]()
Eequation 5-13
![]()
Eequation 5-14
![]()
(xiv) Mass
emission rate in
pounds per hour, (lb/hr).
Eequation 5-15
![]()
Where:
K3 = 63.77 for English units.
(xv) Isokinetic variation using 1 of the following methods:
(A) Calculation from raw data.
Eequation 5-16
![]()
Where:
K4 = 0.003458 mm Hg - m³ml - °K for metric units.
= 0.002672 in. Hg - ft.³/ml - °R for English units.
(B) Calculation from intermediate values.
Eequation 5-17
![]()
Where:
K5 = 4.307 for metric units.
= 0.09409 for English units.
(g) Bibliography:
(i) Federal Register, Volume 42, No. 160, Part 160, Chapter 1, Title 40, Appendix A, Method 5, August 18, 1977.
(ii) Martin, Robert M. Construction Details of Isokinetic Source Sampling Equipment. Environmental Protection Agency. Research Triangle Park, N.C. APTD-0581. April, 1971.
(iii) Rom, Jerome J. Maintenance, Calibration, and Operation of Isokinetic Source Sampling Equipment. Environmental Protection Agency. Research Triangle Park, N.C. APTD-0576. March, 1972.
(iv) Shigehara, R. T. "Adjustments in the EPA Nomograph for Different Pitot Tube Coefficients and Dry Molecular Weights." Stack Sampling News, 2:4 - 11. October, 1974.
(v) Guidelines for Source Testing of Particulate. Michigan Department of Natural Resources, Air Quality Division. June 1, 1977.
R 336.2012 Reference test method 5C.
Rule 1012. Reference test method 5C, out-stack filtration method, reads as follows:
(a) The principle, applicability, and performance test criteria are as follows:
(i) Principle. Particulate matter is
withdrawn iso-kinetically from the source and collected on solid filtering
media maintained at a temperature in the range of 120 ±14 degrees Centigrade,
(248 ±25 degrees Fahrenheit,) or such other another
temperature as specified by the department's rules or a permit condition, or as
approved by the department for a particular application. The particulate mass,
which includes any material that condenses at or above the filtration
temperature, is determined gravimetrically after removal of uncombined water.
(ii) Applicability. This method is applicable for the determination of particulate emissions from stationary sources as identified in table 31 of R 336.1331. The method is also applicable when specifically provided for in the department’s rules, orders, a permit to install, or a permit to operate.
(iii) Performance test criteria as follows:
(A) A performance test mustshall consist meet the
requirements under R 336.2003(2). Of a minimum of 3 separate samples of a
specific air contaminant conducted within a 36-hour period, unless otherwise
authorized by the department. Each of the 3 separate samples shall be obtained
while the source is operating at a similar production level. For the purpose of
determining compliance with an applicable emission limit, rule, or permit
condition, the arithmetic mean of results of the 3 samples shall apply. If a
sample is accidentally lost or conditions occur in which 1 of the 3 samples
must be discontinued because of forced shutdown, failure of an irreplaceable
portion of the sampling train, extreme meteorological conditions, or other circumstances
beyond the owner's or operator's control, compliance may, upon the approval of
the department, be determined using the arithmetic mean of the results of 2
samples.
(B) For any sources that is are subject to an
emission limitation calculated to 50% excess air, the multipoint, integrated
sampling procedure of R 336.2004(1)(c) shall must be used for gas
analysis. For all other sources that require a determination of the molecular
weight of the exhaust, any an optional sampling procedure of R
336.2004(1)(c) may be used. Alternatives or modifications to procedures are
subject to the approval of the department.
(C) The minimum volume per sample shall must be 30 cubic feet
of dry gas corrected to standard conditions, (70 degrees Fahrenheit,
29.92 inches mercury). Minimum sample time shall must be 60 minutes,
which may be continuous or a combination of shorter sampling periods for
sources that operate in a cyclic manner. Smaller sampling times or sample
volumes, when necessitated by process variables or other factors, may be
approved by the department.
(D) For any a source whose
emission control device alters the moisture content of the exhaust gas, a
moisture determination shall must be performed in a location
upstream from the emission control device and in accordance with R
336.2004(1)(d) or an alternative method approved by the department.
(b) The following provisions apply to apparatus:
(i) Sampling train. A schematic of the
sampling train used in this method is shown in figure 103 under R 336.2021.
Construction details for many, but not all, of the train components are given
in APTD-0581, (subdivision
(g)(ii) of this rule). For changes from the APTD-0581 document and for
allowable modifications to figure 103 under R 336.2021, consult with the
department. The operating and maintenance procedures for many, but not all, of
the sampling train are described in APTD-0576, adopted by reference in R
336.1902,
and
referenced under
(subdivision (g)(iii) of this rule). Since correct usage is
important in obtaining valid results, all users shall read APTD-0576 and adopt
the applicable operating and maintenance procedures outlined in it, unless
otherwise specified herein. The sampling train consists of the following
components:
(A) Probe nozzle. Stainless steel (316)
or glass with sharp, tapered leading edge. The angle of taper shall must be less than 30
degrees and the taper shall must be on the outside to preserve a
constant internal diameter. The probe nozzle shall must be of the
buttonhook design, unless otherwise specified by the department. If made of
stainless steel, the nozzle shall must be constructed from seamless
tubing. Other materials of construction may be used, subject to the approval of
the department. A range of nozzle sizes suitable for isokinetic sampling shall must be available, for
example, 0.32 to 1.27 centimeters, (1/8 to 1/2 in.ch,) or larger if
higher volume sampling trains are used inside diameter (ID) nozzles in
increments of 0.16 centimeters, (1/16 in.ches). Each nozzle shall must be calibrated
according to the procedures outlined in subdivision (e) of this
rule.
(B) Probe liner. Borosilicate or
quartz glass tubing with a heating system capable of maintaining a gas
temperature at the exit end during sampling of 120 ±14 degrees Centigrade, (248 ±25
degrees Fahrenheit), another temperature as specified by the department's
rules, or a temperature approved by the department for a particular
application. The tester may opt to operate the equipment at a temperature lower
than that specified. Since the actual temperature at the outlet of the probe is
not usually monitored during sampling, probes constructed according to
APTD-0581,
adopted by reference in R 336.1902, which utilize the calibration curves of
APTD-0576, or calibrated according to the procedure outlined in APTD-0576, adopted by
reference in R 336.1902, are acceptable. Either borosilicate or quartz glass
probe liners may be used for stack temperatures up to about 480 degrees
Centigrade, (900
degrees Fahrenheit); quartz liners shall must be used for
temperatures between 480 and 900 degrees Centigrade, (900 and
1,650 degrees Fahrenheit). Both types of liners may be used at higher
temperatures than specified for short periods of time, subject to the approval
of the department. The softening temperature for borosilicate is 820 degrees
Centigrade, (1,508
degrees Fahrenheit,) and for quartz it
is 1,500 degrees Centigrade, (2,732 degrees Fahrenheit). When
practical, every effort shall must be made to use borosilicate or
quartz glass probe liners. Alternatively, metal liners, such as 316 stainless
steel, Incoloy 825, or other corrosion resistant materials made of seamless
tubing, may be used, subject to the approval of the department.
(C) Pitot tube. Type S, as
described in section 2.1 of method 2, or other another device approved
by the department. The pitot tube shall must be attached to
the probe, as shown in figure 103 under R 336.2021, to allow constant
monitoring of the stack gas velocity. The impact, (high
pressure,) opening plane of
the pitot tube shall must be even with or above the nozzle entry
plane, (see
method 2, figure 2-6b Velocity Traverse Data,) during sampling. The
type S pitot tube assembly shall must have a known coefficient,
determined as outlined in section 4 of method 2.
(D) Differential pressure gauge. Incline
manometer or equivalent devices (2), as described in section 2.2 of
method 2. One manometer shall must be used for velocity head (p) readings,
and the other shall must be used for orifice differential pressure
readings.
(E) Filter holders. Two separate
filter holders in series or 1 filter holder with separate filter supports and
seals for 2 filters. One filter holder with 2 filters held in contact with each
other is not acceptable. Materials of construction may be stainless steel (316),
glass, teflonTeflon, or such other
another material approved
by the department.
(F) Filter heating system. Any heating
system capable of maintaining a temperature around the filter holder during sampling
of 120 ±14 degrees Centigrade, (248 ±25 degrees Fahrenheit),
another temperature as specified by the department's rules or a permit
condition, or a temperature approved by the department for a particular application.
Alternatively, the tester may opt to operate the equipment at a temperature
lower than that specified. A temperature gauge capable of measuring temperature
to within 3 degrees Centigrade, (5.4 degrees Fahrenheit,) shall must be installed so that
the temperature around the filter holders can be regulated and monitored during
sampling. Heating systems other than the one shown in APTD-0581 may be used.
(G) Condenser. The following system shall must be used to
determine the stack gas moisture content: Three impingers connected in series
with leak-free ground glass fittings or any similar leak-free non-contaminating
fittings. All impingers shall must be of the Greenburg-Smith design and
shall
must
be modified by replacing the tip with a 1.3 centimeters, (1/2 in.ch,) ID inside diameter glass tube
extending to about 1.3
centimeters, (1/2 in.ch,) from the bottom of
the flask. Modifications, such as using flexible connections between the
impingers or using materials other than glass, are permitted allowed subject to the approval
of the department’s staff. The first impinger shall must contain a known
quantity of water , (as described in subdivision (d)(i)(C)
of this rule), the second shall must be empty, and the
third shall
must
contain a known weight of silica gel or equivalent desiccant. Alternatively, any
a system that cools
the sample gas stream and allows measurement of the water condensed and
moisture leaving the condenser, each to within 1 milliliter or 1 gram, may be used
subject to the approval of the department. In any case, the means for measuring
the moisture leaving the condenser shall must be by passing the
sample gas stream through a tared silica gel, or equivalent desiccant, trap
with exit gases kept below 20 degrees Centigrade, (68 degrees
Fahrenheit,) and determining the weight gain. If a determination of the
particulate matter collected in the impingers is required by the department's
rules, a permit to install, or a permit to operate, the impinger system
described in this subparagraph shall must be used, without
modification. Contact the department as to the sample recovery and analysis of
the impinger contents.
(H) Metering system. Vacuum gauge,
leak-free pump, thermometers capable of measuring temperature to within 3
degrees Centigrade, (5.4 degrees
Fahrenheit), drygas meter capable of measuring volume to within 2%, and
related equipment as shown in figure 103 under R 336.2021. Other metering
systems capable of maintaining sampling rates within 10% of isokinetic and
capable of determining sample volumes to within 2% may be used, subject to the
approval of the department. When the metering system is used in conjunction with
a pitot tube, the system shall must enable checks of isokinetic rates.
Sampling trains utilizing metering systems designed for higher flow rates than those
described in APTD-0581 or APTD-0576, both adopted by reference in R 336.1902, may be used if the
specifications of this method are met.
(I) Barometer. Mercury, aneroid, or other
barometer capable of measuring atmospheric pressure to within 2.5 millimeters Hgmercury, (0.1 in.ch Hgmercury). In many cases, the
barometric reading may be obtained from a nearby national weather service
station. When obtained from this source, the station value, which is the
absolute barometric pressure, shall must be requested and an
adjustment for elevation differences between the weather station and sampling
point shall
must
be applied at a rate of minus 2.5 millimeters Hgmercury per 30
meters,
(0.1 in.ch Hgmercury,) per 30 m (100
foot.), elevation
increase or vice versa for elevation decrease.
(J) Gas density determination equipment.
Temperature sensor and pressure gauge, as described in sections 2.3 and 2.4
of method 2, and gas analyzer, if necessary, as described in method 3. The temperature
sensor shall
must,
preferably, be permanently attached to the pitot tube or sampling probe in a
fixed configuration such so that the tip of the sensor extends beyond
the leading edge of the probe sheath and does not touch any metal.
Alternatively, the sensor may be attached just prior to before use in the field.
Note, however, that if the temperature sensor is attached in the field, the
sensor shall
must
be placed in an interference-free arrangement with respect to the type S pitot
tube openings, (see method
2, figure 2.7). As a second alternative, if a difference of not more
than 1% in the average velocity measurement is to be introduced, the
temperature gauge need not be attached to the probe or pitot tube. This
alternative is subject to the approval of the department.
(ii) Sample recovery. The following
items are needed:
(A) Probe-liner and probe-nozzle brushes.
Nylon bristle brushes with stainless steel wire handles. The probe brush shall must have extensions, at
least as long as the probe, made of stainless steel, nylon, teflonTeflon, or similarly
inert material. The brushes shall must be properly sized and shaped to
brush out the probe liner and nozzle.
(B) Wash bottles--2bottles - 2. Glass wash
bottles are recommended; polyethylene wash bottles may be used at the option of
the tester. It is recommended that acetone not be stored in polyethylene
bottles for longer than a month.
(C) Glass sample storage containers. Chemically
resistant, borosilicate glass bottles, for acetone washes, 500 ml or 1000 milliliters. Screw cap liners
shall
must
either be rubber-backed teflonTeflon or shall must be constructed so
as to be leak-free and resistant to chemical attack by acetone. Narrow-mouth glass
bottles have been found to be less prone to leakage. Alternatively,
polyethylene bottles may be used.
(D) Filter containers. Glass, polyethylene, or aluminum tube containers, unless otherwise specified by the department.
(E) Graduated cylinder or balance. To
measure condensed water to within 1 milliliter or 1 gram. Graduated
cylinders shall
must
have subdivisions of not more than 2 milliliters. Most laboratory
balances are capable of weighing to the nearest 0.5 gram or less. Any of
these balances are suitable for use here and in paragraph (iii)(D) of this
subdivision.
(F) Plastic storage containers. Airtight containers to store silica gel.
(G) Funnel and rubber policeman. T, to aid in the
transfer of silica gel to container; not necessary if silica gel is weighed in
the field.
(H) Funnel. made from Gglass or
polyethylene, to aid in sample recovery.
(iii) Analysis. must include Tthe following
equipment is needed for analysis:
(A) Glass weighing dishes.
(B) Desiccator.
(C) Analytical balance., Tto measure to
within 0.1
milligrams.
(D) Balance., Tto measure to
within 0.5
milligrams.
(E) Beakers., 250 milliliters.
(F) Hygrometer., Tto measure the relative
humidity of the laboratory environment.
(G) Temperature gauge., Tto measure the temperature
of the laboratory environment.
(c) The following provisions apply to reagents:
(i) Sampling. The reagents used
in sampling are as follows:
(A) Filters. Two outstack filters may
be any combination of alundum ceramic thimble filters, type RA-98 or glass
fiber filters, type A without organic binder. The size of such the filters shall must allow proper sampling
rates to maintain isokinetics using the nozzle sizes specified in subdivision
(b)(i)(A) of this rule. Alternatively, other types of filters may be used,
subject to the approval of the department.
(B) Silica gel. Indicating type, 6 to
16 mesh. If previously used, dry at 175 degrees Centigrade, (350 degrees
Fahrenheit), for 2 hours. New
silica gel may be used as received. Alternatively, other types of desiccants, (equivalent
or better,) may be used,
subject to the approval of the department.
(C) Water. When analysis of the material
caught in the impingers is required, distilled water shall must be used. Run
blanks prior to field use to eliminate a high blank on test samples.
(D) Crushed ice.
(E) Stopcock grease.
Acetone-insoluble, heatstable silicone grease. This is not necessary if screw
on connectors with teflonTeflon sleeves, or equivalent, are used.
Alternatively, other types of stopcock grease may be used, subject to the
approval of the department.
(ii) Sample recovery., Wwashing solvent.
Either acetone or distilled water may be used for sample recovery. If acetone
is used for washing solvent, then reagent grade, less than 0.001% residue, in glass
bottles is required. Acetone from metal containers generally has a high residue
blank and shall
must
not be used. Suppliers sometimes transfer acetone to glass bottles from metal containers;
thus, so acetone blanks shall must be run prior
to before field use and only
acetone with low blank values, (less than 0.001%,) shall must be used. A blank
value of more than 0.001% of the weight of acetone used shall must not be subtracted
from the sample weight. If distilled water is used for washing solvent, use
distilled water with less than 0.001% residue. Run blanks before field use to
eliminate a high blank on test samples.
(iii) Analysis. Two reagents
are required for the analysis:
(A) Solvent. Same as paragraph (ii) of this subdivision for quantitative transfer.
(B) Desiccant. Anhydrous calcium sulfate, indicating type. Alternatively, other types of desiccants may be used, subject to the approval of the department.
(d) The following provisions apply to procedure:
(i) Sampling. The complexity of this
method is such that, in order to obtain reliable results, testers shall be
trained and experienced with the test procedures. Sampling shall must comply with the
following provisions:
(A) Pretest preparation. All the components
shall must be maintained and calibrated according to the applicable
procedures described in APTD-0576, adopted by reference in R 336.1902,
unless otherwise specified in this rule. Weigh several 200 to 300 gram
portions of silica gel in airtight containers to the nearest 0.5 gram.
Record the total weight of the silica gel plus container on each container. As
an alternative, the silica gel need not be pre-weighed, but may be weighed
directly in its impinger or sampling holder just prior to before train
assembly. Check filters visually against light for irregularities, flaws, pinhole
leaks, or cracks. Label filters of the proper size on the back side using
numbering machine ink. As an alternative, label the shipping containers,
described in (subdivision (b)(ii)(D) of this rule,) and
keep the filters in these containers at all times, except during sampling and
weighing. Dry the filters in an oven at 105 degrees Centigrade, (220
degrees Fahrenheit), for a minimum of 2 hours, cool for at least 1
hour in a desiccator containing anhydrous calcium sulfate, and individually weigh
and record their weights to the nearest 0.1 milligram.
During the weighing, the filters shall must not be exposed to the
laboratory atmosphere for a period of more than 2 minutes and a relative
humidity above 50%. Procedures, other than those specified, that account for
relative humidity effects may be used, subject to the approval of the
department.
(B) Preliminary determinations.
Select the sampling site and the minimum number of sampling points according to
method 1 or as specified by the department. Determine the stack pressure, temperature,
and the range of velocity heads using method 2.; it It is recommended that
a leak check of the pitot lines, (see method 2, section 3.1), be performed.
Determine the moisture content using approximation method 4, or its
alternatives, for the purpose of making isokinetic sampling rate settings.
Determine the stack gas dry molecular weight, as described in method 2., section 3.6; if If integrated method
3 sampling is used for molecular weight determination, the integrated bag sample
shall
must
be taken simultaneously with, and for the same total length of time as, the
particulate sample run. Select a nozzle size based on the range of velocity
heads so that it is not necessary to change the nozzle size in order to
maintain isokinetic sampling rates. During the run, do not change the nozzle
size. Ensure that the proper differential pressure gauge is chosen for the range
of velocity heads encountered, (see section 2.2 of method
2). Select a suitable probe liner and probe length so that all traverse points
can be sampled. For large stacks, consider sampling from opposite sides of the stack
to reduce the length of probes. Select a total sampling time greater than or equal
to the minimum total sampling time specified in the test procedures for the
specific industry so that the sampling time per point is not less than 5
minutes, unless approved by the department, or some greater time interval as specified
by the department, and so that the sample volume taken, corrected to standard
conditions, exceeds the required minimum total gas sample volume. The latter is
based on an approximate average sampling rate. It is recommended that the number
of minutes sampled at each point be an integer or an integer plus 1/2 minute to
avoid timekeeping errors. In some circumstances, such as in batch cycles, it
may be necessary to sample for shorter times at the traverse points and to obtain
smaller gas sample volumes. In these cases, the department's approval shall must first be
obtained.
(C) Preparation of collection train.
During preparation and assembly of the sampling train, keep all openings where
contamination can occur covered until just before assembly or until sampling is
about to begin. Place 100 milliliters of water in the first
impinger, leave the second impinger empty, and transfer approximately 200 to
300 grams of pre-weighed silica gel from its container to the
third impinger. More silica gel may be used, but care should be taken to ensure
that it is not entrained and carried out from the impinger during sampling.
Place the container in a clean place for later use in the sample recovery.
Alternatively, the weight of the silica gel plus impinger may be determined to
the nearest 0.5 gram and recorded. Using tweezers or clean disposable
surgical gloves, place a labeled, (identified), and
weighed filter in the filter holder. Be sure that the filter is properly
centered and the gasket properly placed so as to prevent the sample gas stream
from circumventing the filter. Check the filter for tears after assembly is
completed. When glass liners are used, install the selected nozzle using a
Viton A O-0ring when stack temperatures are less than 260 degrees
Centigrade, (500 degrees Fahrenheit), and an asbestos
string gasket when temperatures are higher. See APTD-0576, adopted by
reference in R 336.1902, for details. Other connecting systems using either
310 stainless steel or teflon Teflon ferrules may be used. When
metal liners are used, install the nozzle in the same manner as for glass liners
or by a leak-free direct mechanical connection. Mark the probe with
heat-resistant tape or by some other method to denote the proper distance into
the stack or duct for each sampling point. Set up the train as in figure 103
under R 336.2021. If necessary, use a very light coat of silicone grease on
all ground glass joints. Grease only the outer portion, (see
APTD-0576, adopted by reference in R 336.1902,) to avoid the
possibility of contamination by the silicone grease. Place crushed ice around
the impingers.
(D) Leak check procedures as follows:
(I1) Pretest leak
check. A pretest leak check is strongly recommended, but not required, to
prevent invalid sampling and wasted time. If the tester opts to conduct the
pretest leak check, the following procedure shall must be used: After
the sampling train has been assembled, turn it on and set the filter and probe
heating systems at the desired operating temperatures. Allow time for the
temperatures to stabilize. If a Viton A O0-ring or other leak-free connection
is used in assembling the probe nozzle to the probe liner, leak check the train
at the sampling site by plugging the nozzle and pulling a 380 millimeter mercury, Hg (15 inch. Hgmercury,) vacuum. A lower
vacuum may be used, if it is not exceeded during the test. If an asbestos
string is used, do not connect the probe to the train during the leak check. Instead,
leak check the train by first plugging the inlet to the filter holder, (and cyclone, if
applicable,) and pulling a 380 millimeter Hgmercury, (15 inch. Hgmercury), vacuum. A lower
vacuum may be used if it is not exceeded during the test. Then connect the
probe to the train and leak check at about a 25 millimeter Hgmercury, (1 inch. Hgmercury,) vacuum.; Alternativelyalternatively, the probe may be
leak checked with the rest of the sampling train, in 1 step, at a 380 millimeter Hgmercury, (15 inch. Hgmercury,) vacuum. Leakage
rates in excess of 4% of the average sampling rate or 0.00057 cubic meters per
minute
m³/min (0.02 cubic
feet per minutecfm), whichever is
less, are unacceptable. The following leak check instructions for the sampling train
described in APTD-0576 and APTD-0581 may be helpful. Start the pump with the
bypass valve fully open and the coarse adjust valve completely closed.
Partially open the coarse adjust valve and slowly close the bypass valve until the
desired vacuum is reached. Do not reverse the direction of the bypass valve,; as this will cause
water to back up into the filter holder. If the desired vacuum is exceeded,
either leak check at this higher vacuum or end the leak check and start over.
When the leak check is completed, first slowly remove the plug from the inlet
to the probe, filter holder, or cyclone, (if applicable), and immediately
turn off the vacuum pump. This prevents the water in the impingers from being
forced backward into the filter holder and prevents silica gel from being
entrained backward into the third impinger.
(II2) Leak checks
during sample run. If, during the sampling run, a component, (such as a
filter assembly or impinger), change becomes necessary, a leak check shall must be conducted
immediately before the change is made. The leak check shall must be done according
to the procedure outlined in paragraph (i)(D)(1I) of this
subdivision
subparagraph
(D) (I) of this paragraph, except that it shall must be done at a vacuum
equal to or greater than the maximum value recorded up to that point in the
test. If the leakage rate is found to be not more than 0.00057 cubic meters per
minute,
m³/min (0.02 cubic
feet per minute,cfm) or 4% of the average
sampling rate, whichever is less, the results are acceptable and no correction
need be applied to the total volume of dry gas metered;. Iif, however, a higher
leakage rate is obtained, the tester shall either record the leakage rate and
plan to correct the sample volume, as shown in subdivision (f)(iii) of
R
336.2011this
rule,
or shall void the sampling run. Immediately after component changes, leak checks
are optional;.
If the if
such
leak checks are done, the procedure outlined in paragraph (i)(D)(1I) of this
subdivision shall must be used.
(III3) Post-test leak
check. A leak check is mandatory at the conclusion of each sampling run. The
leak check shall must be done in accordance with the procedures
outlined in paragraph (i)(D)(1I) of this subdivision, except that it shall must be conducted at a
vacuum equal to or greater than the maximum value reached during the sampling
run. If the leakage rate is found to be not more than 0.00057 m³/mincubic meters per
minute,
(0.02 cfmcubic feet per minute,) or 4% of the average
sampling rate, whichever is less, the results are acceptable and no correction need
be applied to the total volume of dry gas metered. If, however, a higher leakage
rate is obtained, the tester shall either record the leakage rate and correct the
sample volume, as shown in subdivision (f)(iii) of R 336.2011this rule, or shall void
the sampling run.
(E) Particulate train operation. During
the sampling run, maintain an isokinetic sampling rate that is within 10% of true
isokinetic, unless otherwise specified by the department. For each run, record
the data required on a data sheet such as the one shown in figure 104 under
R 336.2021. Be sure to record the initial dry-gas meter reading. Record the
dry-gas meter readings at the beginning and end of each sampling time
increment, when changes in flow rates are made, before and after each leak
check, and when sampling is halted. Take other readings required by figure 104
under R 336.2021 at least once at each sample point during each time
increment, and take additional readings when significant changes, (20%
variation in velocity head readings), necessitate additional adjustments
in flow rate. Level and zero the manometer. Because the manometer level and
zero may drift due to vibrations and temperature changes, make periodic checks
during the traverse. Clean the portholes prior to before the test run to
minimize the chance of sampling deposited material. To begin sampling, remove
the nozzle cap and verify that the pitot tube and probe are properly
positioned. Position the nozzle at the first traverse point with the tip
pointing directly into the gas stream. Immediately start the pump and adjust the
flow to isokinetic conditions. Nomographs that aid in the rapid adjustment of
the isokinetic sampling rate without excessive computations are available.
These nomographs are designed for use when the type S pitot tube coefficient is
0.85 ±0.02 and the stack gas equivalent density, (dry
molecular weight,) is equal to 29 ±4.
APTD-0576,
adopted by reference in R 336.1902, details the procedure for using the
nomographs. If Cp and Md are outside the above stated ranges, do not use the
nomographs unless appropriate steps, (see
subdivision (g)(iv) of this rule,) are taken to compensate for the
deviations. When the stack is under significant negative pressure, (height of
impinger stem), take care to pull low flow when inserting the probe into
the stack to prevent water from backing into the sample tubing and to avoid pulsation
through the filter and possible loss of materials. When the probe is in
position, block off the openings around the probe and porthole to prevent
unrepresentative dilution of the gas stream. Traverse the stack cross section,
as required by method 1 or as specified by the department, being careful not to
bump the probe nozzle into the stack walls when sampling near the walls or when
removing or inserting the probe through the portholes.; this This minimizes the chance
of extracting deposited material. During the test run, add more ice and, if necessary,
salt to maintain a temperature of less than 20 degrees Centigrade, (68 degrees Fahrenheit,) at the condenser/silica
gel outlet. Also, periodically check the level and zero of the manometer. If
the pressure drop across the filter becomes too high and makes isokinetic
sampling difficult to maintain, the filter may be replaced in the midst of a
sample run. It is recommended that another complete filter assembly be used
rather than attempting to change the filter itself. Before a new filter
assembly is installed, conduct a leak check, (see subparagraph paragraph (i)(D)(2II) of this paragraph subdivision).
The total particulate weight shall must include the summation of all
filter assembly catches. A single train shall must be used for the
entire sample run, except in cases where simultaneous sampling is required in 2
or more separate ducts, at 2 or more different locations within the same duct, or
where equipment failure necessitates a change of trains. In all other
situations, the use of 2 or more trains shall must be subject to the
approval of the department. Note that when 2 or more trains are used, separate
analyses of the front half catches from the individual trains may be combined, as
may the impinger catches, and 1 analysis of the front half catch and 1 analysis
of impinger catch may be performed. Consult with the department for details concerning
the calculation of results when 2 or more trains are used. At the end of the
sample run, turn off the coarse adjust valve, remove the probe and nozzle from
the stack, turn off the pump, record the final dry gas meter reading, and
conduct a post-test leak check, as outlined in paragraph subparagraph (i)(D)(3III) of this paragraph. Also, leak check
the pitot lines as described in method 2, section 3.1; the. The lines shall must pass this leak
check to validate the velocity head data.
(F) Calculation of percent isokinetic.
Calculate percent isokinetic, (see subdivision (f) of this rule,) to determine
whether the run was valid or whether another test run should be made. If there was
difficulty in maintaining isokinetic rates due to source conditions, consult
with the department for possible variance on the isokinetic rates.
(ii) Sample recovery. Proper cleanup
procedure begins as soon as the probe is removed from the stack at the end of
the sampling period. Allow the probe to cool. When the probe can be safely handled,
wipe off all external particulate matter near the tip of the probe nozzle and
place a cap over it to prevent losing or gaining particulate matter. Do not cap
off the probe tip tightly while the sampling train is cooling down as this creates
a vacuum in the filter holder and draws water from the impingers into the
filter holder. Before moving the sample train to the cleanup site, remove the
probe from the sample train, wipe off the silicone grease, and cap the open outlet
of the probe. Be careful not to lose any condensate that might be present. Wipe
off the silicone grease from the filter inlet where the probe was fastened and
cap it. Remove the umbilical cord from the last impinger and cap the impinger.
If a flexible line is used between the first impinger or condenser and the
filter holder, disconnect the line at the filter holder and let any condensed
water or liquid drain into the impingers or condenser. After wiping off the
silicone grease, cap off the filter holder outlet and impinger inlet.
Ground-glass stoppers, plastic caps, or serum caps may be used to close these
openings. Transfer the probe and filter-impinger assembly to the cleanup area. This
area shall
must
be clean and protected from the wind so that the chances of contaminating or
losing the sample are minimized. Save a portion of the solvent used for cleanup
as a blank. Take 200
milliliters of this solvent
directly from the wash bottle being used and place it in a glass sample
container labeled "solvent blank." Inspect the train prior to and
during disassembly and note any abnormal conditions. Treat the samples
as follows:
(A) Container Nos.numbers
1, 1A. Carefully remove the filters from the filter holders and place in their identified
containers. Use a pair of tweezers or clean disposable surgical gloves, or both,
to handle the filters. Carefully transfer to the container any particulate
matter or filter fibers, or both, that adhere to the filter holder gasket by
using a dry nylon bristle brush or sharp-edged blade, or both. Seal the
container.
(B) Container No.number 2. Taking care to
see that dust on the outside of the probe or other exterior surfaces does not get
into the sample, the testoer shall quantitatively recover from
particulate matter or any condensate from the nozzle, probe fitting, probe
liner, and from both filter holders by washing these components with solvent
and placing the wash in a glass container. Perform the solvent rinses as follows:
Carefully remove the probe nozzle and clean the inside surface by rinsing with
solvent from a wash bottle and brushing with a nylon bristle brush. Brush until
the solvent rinse shows no visible particles and then make a final rinse of the
inside surface with solvent. Brush and rinse the inside parts of the Swagelok
fitting with solvent in a similar way until no visible particles remain. Rinse
the probe liner with solvent by tilting and rotating the probe while squirting
solvent into its upper end so that all inside surfaces are wetted with acetone.
Let the solvent drain from the lower end into the sample container. A glass or
polyethylene funnel may be used to aid in transferring liquid washes to the
container. Follow the solvent rinse with a probe brush. Hold the probe in an
inclined position and squirt solvent into the upper end as the probe brush is
being pushed with a twisting action through the probe.; hold Hold a sample
container underneath the lower end of the probe and catch any solvent and particulate
matter that is brushed from the probe. Run the brush through the probe 3 or
more times until no visible particulate matter is carried out with the solvent
or until none remains in the probe liner on visual inspection. With stainless
steel or other metal probes, run the brush through, in the above prescribed
manner, not less than 6 times, since metal probes have small crevices in which particulate
matter can be entrapped. Rinse the brush with solvent and quantitatively
collect these washings in the sample container. After the brushing, make a
final solvent rinse of the probe as described above. It is recommended that 2
people be used to clean the probe to minimize sample losses. Between sampling
runs, keep brushes clean and protected from contamination. After ensuring that
all joints have been wiped clean of silicone grease, clean the inside of both
filter holders by rubbing the surfaces with a nylon bristle brush and rinsing
with solvent. Rinse each surface 3 times, or more if needed, to remove visible
particulate. Make a final rinse of the brush and filter holder. After all
solvent washings and particulate matter have been collected in the sample
container, tighten the lid on the sample container so that solvent will not
leak out when it is shipped to the laboratory. Mark the height of the fluid level
to determine whether or not leakage occurred during transport. Label the
container to clearly identify its contents.
(C) Container No.number 3. Note the color
of the indicating silica gel to determine if it has been completely spent and
make a notation of its condition. Transfer the silica gel from the third
impinger to its original container and seal. A funnel may make it easier to
pour the silica gel without spilling it. A rubber policeman may be used as an
aid in removing the silica gel from the impinger. It is not necessary to remove
the small amount of dust particles that adhere to the impinger wall and are
difficult to remove. Since the gain in weight is to be used for moisture
calculations, do not use any water or other liquids to transfer the silica gel.
If a balance is available in the field, follow the procedure for container No.number 3 in paragraph (iii)(C) of this
subdivision. Impinger water. Treat the impingers as follows: Make a notation of
any color or film in the liquid catch. Measure the liquid that is in the first 2
impingers to within ±1 milliliter by using a graduated cylinder or
by weighing it to within ±1.0 g gram by using a balance if none is
available. Record the volume or weight of liquid present. This information is required
to calculate the moisture content of the effluent gas. Discard the liquid after
measuring and recording the volume or weight, unless analysis of the impinger
catch is required, (see
subdivision (b)(i)(G) of this rule). If a different type of condenser is
used, measure the amount of moisture condensed either volumetrically or
gravimetrically. Whenever possible, containers shall must be shipped in a
manner that keeps them upright at all times.
(iii) Analysis. Record the data required on a sheet such as the one shown in figure 106 under R 336.2021. Handle each sample container as follows:
(A) Container Nos.numbers 1, 1A. Analyze
and report each filter separately. Transfer the filter and any loose
particulate from the sample container to a tared-glass weighing dish. Dry the filter
in an oven at 105 degrees Centigrade, (220 degrees Fahrenheit,) for a minimum of
2 hours, cool for at least 1 hour in a desiccator containing anhydrous calcium
sulfate, and weigh and record its weight to the nearest 0.1 milligram. During the weighing,
the filter shall must not be exposed to the laboratory
atmosphere for a period of more than 2 minutes or a relative humidity above
50%. Procedures, other than those specified, that account for relative humidity
effects may be used, subject to the approval of the department. The method used
for the drying and weighing of filters shall must be consistent
before and after the test.
(B) Container No.number 2. Note the level
of liquid in the container and confirm on the analysis sheet whether or not leakage
occurred during transport. If a noticeable amount of leakage has occurred,
either void the sample or use methods, subject to the approval of the department,
to correct the final results. Measure the liquid in this container either volumetrically
to ±1
milliliter or
gravimetrically to ±1.0 gram. Transfer the contents to a tared 250-
milliliter beaker and
evaporate to dryness either at ambient temperature and pressure for acetone or
at 95 degrees Centigrade, (203 degrees Fahrenheit,) in an oven for
distilled water. Then subject the sample to 250 degrees Centigrade, (482 degrees
Fahrenheit,) in an oven for 2
to 3 hours. Desiccate for 24 hours and weigh to a constant weight. Report the
results to the nearest 0.1 milligram.
(C) Container No.number 3. Weigh the
spent silica gel, or silica gel plus impinger, to the nearest 0.5 gram using a balance.
This step may be conducted in the field. "Solvent blank" container. Measure
solvent in this container either volumetrically or gravimetrically. Transfer the
contents to a tared 250- milliliter beaker and
evaporate to dryness either at ambient temperature and pressure for acetone or
at 95 degrees Centigrade, (203 degrees Fahrenheit), in an oven for
distilled water. Then subject the sample to 250 degrees Centigrade, (482 degrees
Fahrenheit), in an oven for 2
to 3 hours. Desiccate for 24-hours and weigh to a constant weight. Report the
results to the nearest 0.1 milligram. If acetone is used, the contents of
container No.number 2, as well as the
acetone blank container, may be evaporated at temperatures higher than ambient.
If evaporation is done at an elevated temperature, the temperature shall must be closely supervised,
and the contents of the beaker shall must be swirled
occasionally to maintain an even temperature. Use extreme care, as acetone is
highly flammable and has a low flash point.
(e) Calibration. Maintain a laboratory log
of all calibrations. Calibrations shall must comply with all
of the following provisions:
(i) Probe nozzle. Probe nozzles shall must be calibrated
before their initial use in the field. Using a micrometer, measure the inside
diameter of the nozzle to the nearest 0.025 millimeter, (0.001 in.ch). Make 3 separate measurements
using different diameters each time and obtain the average of the measurements.
The difference between the high and low numbers shall must not exceed 0.1 millimeter, (0.004 in.ch). When nozzles
become nicked, dented, or corroded, they shall must be reshaped,
sharpened, and recalibrated before use. Each nozzle shall must be permanently
and uniquely identified.
(ii) Pitot tube. The type S pitot tube
assembly shall
must
be calibrated according to the procedure outlined in section 4 of method
2.
(iii) Metering system. Before its initial
use in the field, the metering system shall must be calibrated
according to the procedure outlined in APTD -0576, adopted by reference
in R 336.1902.
Instead of physically adjusting the dry gas meter dial readings to correspond
to the wet test meter readings, calibration factors may be used to
mathematically correct the gas meter dial readings to the proper values. Before
calibrating the metering system, it is suggested that a leak check be
conducted. For metering systems having diaphragm or rotary pumps, the normal
leak check procedure will not detect leakages within the pump. For these cases,
the following leak check procedure is suggested: Make a 10-minute calibration
run at 0.00057 cubic
meters per minute, m³/min (0.02 cubic feet per minute. cfm); at At the end of the
run, take the difference of the measured wet test meter and dry gas meter volumes, and; divide the difference
by 10 to get the leak rate. The leak rate shall must not exceed
0.00057 cubic
meters per minute, m³/min (0.02 cubic feet per minutecfm). After each field
use, the calibration of the metering system shall must be checked by
performing 3 calibration runs at a single, intermediate orifice setting, based
on the previous field test, with the vacuum set at the maximum value reached
during the test series. To adjust the vacuum, insert a valve between the wet
test meter and the inlet of the metering system. Calculate the average value of
the calibration factor. If the calibration has changed by more than 5%,
recalibrate the meter over the full range of orifice settings, as outlined in
APTD-0576. Alternatively, a spirometer may be substituted for a wettest meter in
the above mentioned calibration procedures. Alternative procedures, such as
using the orifice meter coefficients, may be used, subject to the approval of
the department. If the dry gas meter coefficient values obtained before and after
a test series differ by more than 5%, the test series shall must be performed using
whichever meter coefficient value, (before or after,) gives the lower
value of total sample volume.
(iv) Probe heater calibration. The probe
heating system shall must be calibrated before its initial use in
the field according to the procedures outlined in APTD-0576, adopted by
reference in R 336.1902. Probes constructed according to APTD-0581 need not
be calibrated if the calibration curves in APTD-0576 are used.
(v) Temperature gauges. Use the
procedure in section 4.3 of method 2 to calibrate in stack temperature
gauges. Dial thermometers, such as those used for the dry gas meter and condenser
outlet, shall
must
be calibrated against mercury in glass thermometers.
(vi) Leak check of metering system
shown in figure 103 under R 336.2021. That portion of the sampling train
from the pump to the orifice meter shall must be leak checked
before initial use and after each shipment. Leakage after the pump will
resultresults in less volume
being recorded than is actually sampled. The following procedure is suggested, also (see
figure 107 under R 336.2021): Close the main valve on the meter
box. Insert a 1-hole rubber stopper with rubber tubing attached into the
orifice exhaust pipe. Disconnect and vent the low side of the orifice
manometer. Close off the low side orifice tap. Pressurize the system to 13 to
18 centimeters, (5 to 7 in.ches,) water column by
blowing into the rubber tubing. Pinch off the tubing and observe the manometer
for 1 minute. A loss of pressure on the manometer indicates a leak in the meter
box.; Leaksleaks, if present, shall must be corrected.
(vii) Barometer. Calibrate against a mercury barometer.
(f) Calculations. When carrying out
calculations, retain at least 1 extra decimal figure beyond that of the acquired
data. Round off figures after the final calculation. Other forms of the
equations may be used if they the other forms of the equations give equivalent
results. All of the following provisions under R 336.2011
(f) apply
to calculations: for this rule.
An =
Cross-sectional area of nozzle, m²(ft.²).
A =
Cross-sectional area of stack or flue at the point of sampling, ft².
B ws =
Water vapor in the gas stream, proportion by volume, expressed as a fraction.
B wi = Percent water vapor in
gas entering source particulate control device determined by method 4.
B wo =
Percent water vapor in gas exiting source particulate control device.
Ca =
Wash blank residue concentration, mg/g.
Cs = Concentration of
particulate matter in stack gas, pounds per 1,000 pounds of actual stack gas.
C sD = Concentration of
particulate matter in stack gas, moisture excluded, pounds per 1000 pounds of
dry stack gas.
Cs50 = Concentration of
particulate matter corrected to 50% excess air, pounds per 1000 pounds of stack
gas.
Cs50D = Concentration of particulate
matter corrected to 50% excess air, excluding any water addition from a
collector, pounds per 1000 pounds of stack gas.
E = Mass emission rate of particulate,
lb/hr.
F50 = Concentration
conversion factor to 50% excess air with no moisture alterations in exhaust.
F50D = Concentration
conversion factor to 50% excess air, excluding any moisture added to exhaust
gas by pollution collection system.
FD = Concentration conversion factor to dry basis, excluding any
water in the stack gas.
I = Percent of isokinetic sampling.
L a = Maximum acceptable leakage rate for either a pretest leak
check or for a leak check following a component change; equal to 0.00057 m³/min
(0.02 cfm) or 4% of the average sampling rate, whichever is less.
Li = Individual leakage rate observed during the leak check
conducted
before
the "ith" component change (i = 1, 2, 3 . . . . n), m³/min (cfm).
Lp = Leakage rate observed during the post-test leak check, m³/min
(cfm).
Md = Molecular weight of dry stack gas, g/g mole (lb/lb-mole),
calculated
by
method 3, equation 3-21, using data from integrated method
3.
mn = Total amount of particulate matter collected, mg.
Mw = Molecular weight of water, 18.0 g/g-mole (18.0 lb/lb-mole).
ma = Mass of residue of solvent after evaporation, mg.
mg = Total weight of gas samples through nozzle, lb.
P bar = Barometric pressure at the sampling site, mm Hg (in. Hg).
Ps = Absolute stack gas pressure.
Pstd = Standard absolute pressure, 760 mm Hg (29.92 in. Hg).
R = Ideal gas constant, 0.06236 mm Hg-m³/°K-g-mole (21.85
in.Hg-ft.³/°R?lb-mole).
T m = Absolute average dry-gas meter temperature (see figure 104),
°K (°R).
Ts = Absolute average stack gas temperature (see figure 104), °K
(°R).
Tstd = Standard absolute temperature, 294.I°K (530°R).
V a = Volume of solvent blank, ml.
V aw = Volume of solvent used in wash, ml.
V lc = Total volume of liquid collected in impingers and silica
gel (see figure 106), ml.
Vm = Volume of gas sample as measured by the dry-gas meter, dcm
(dcf).
V m(std) = Volume of gas sample measured by the dry-gas meter,
corrected to standard conditions, dscm (dscf).
V w(std) = Volume of water vapor in the gas sample, corrected to
standard conditions, scm (scf).
V s = Stack gas velocity, calculated by method 2, equation 2-9,
using data obtained from method 5, m/sec (ft./sec).
Wa = Weight of residue in solvent wash, mg.
Y = Dry-gas meter calibration factor.
ΔH = Average pressure differential across the orifice meter (see figure
104), mm H20 (in. H20).
%02 = Percent oxygen in stack gas by volume (dry basis).
%N2 = Percent nitrogen in stack gas by volume (dry basis).
p a = Density of solvent, mg/ml.
p s(std) = Density of all sampled gas at standard conditions,
lb/ft.³
pw = Density of water, 0.9982 g/ml (0.002201 lb/ml).
θ = Total sample time, min.
θ1 = Sample time, interval, from the beginning of a run until the first
component change, min.
θi = Sampling time interval, between 2 successive component changes,
beginning with the interval between the first and second changes, min.
θp = Sampling time interval, from the final (nth) component change until
the end of the sampling run, min.
13.6 = Specific gravity of mercury.
60 = Sec/min.
100 = Conversion to percent.
386.9 = Cubic feet per lb-mole of ideal gas at standard conditions.
453.6 = Conversion of pounds to grams.
3600 = Conversion of hours to sec.
1000 = Conversion of 1000 lb units to lb units.
(ii) Average the dry gas meter
temperature and average the orifice pressure drop. See data sheet (figure 104).
(iii) Dry gas volume. Correct the
sample volume measured by the dry gas meter to standard conditions (21.1
degrees Centigrade, 760 mm Hg or 70 degrees Fahrenheit,
29.92 in. Hg) by using equation 5-1.
|
|
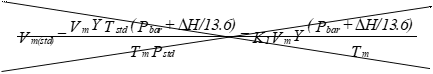
Where:
K1 = 0.3869 °K/mm Hg for metric units.
= 17.71 °R/in.
Hg for English units.
Equation 5-1 can
be used as written. However, if the leakage rate observed during any of the
mandatory leak checks (for example, the post-test leak check or leak checks
conducted prior to component changes) exceeds La, equation 5-1 shall be modified
as follows:
(A) Case I.
No component changes made during sampling run. In this case, replace Vm
in equation 5-1 with the expression:
![]()
(B) Case II.
One or more component changes made during the sampling run. In this case,
replace Vm in equation 5-1 by
the expression:
![]()
and substitute only for those leakage
rates (Li or Lp) that exceed La.
(iv) Volume of water vapor.
|
|
Where:
K2=0.001338 m³/ml for metric units.
=0.04733 ft.³/ml for English units.
(v) Moisture content.
|
|
In saturated or
water droplet-laden gas streams, 2 calculations of the moisture content of the
stack gas shall be made:1 from the impinger analysis (equation 5-3), and a
second from the assumption of saturated conditions. The lower of the 2 values
of Bws shall be considered correct. The procedure for determining
the moisture content based upon assumption of saturated conditions is given in
the note of section 1.2 of method 4. For the purpose of this method, the
average stack gas temperature from figure 104 under R 336.2021 may be
used to make the determination, if the accuracy of the in-stack temperature
sensor is ±1 degree Centigrade (2 degrees Fahrenheit).
(vi) Solvent
blank concentration.
|
|
(vii) Solvent
wash blank.
|
|
(viii) Total particulate
weight. Determine the total particulate catch from the sum of the weights
obtained from containers 1, 1A, and 2 less the wash solvent blank (see figure
106).
Refer to
subdivision (d)(i)(E) of this rule to assist in the calculation of results involving
2 or more pairs of filters or 2 or more sampling trains.
(ix) Sampled
gas density. Determine the density of the gas sampled from the stack, at
standard conditions (lb/ft.³).
|
|
(x) Total
weight of gas sampled (lbs).
|
|
(xi) Particulate
concentration (lbs/1000 lbs).
|
|
(xii) Excess
air and moisture correction factors:
(A) Correction
factor to 50% excess air for those sources with or without any particulate
collector where no increase in moisture content of the exhaust gas occurs after
the process and before the point of sampling.
|
|
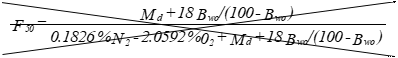
(B) Correction
factor to 50% excess air for those sources with a wet collection device
(scrubber) that increases the moisture content of the exhaust gas after the
process and before the point of sampling.
equation 5-10
|
|
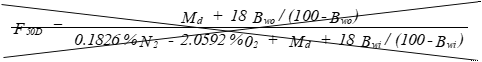
(C) Correction
factor to convert the actual concentration, Cs, to dry conditions.
equation 5-11
|
|
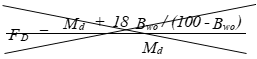
(xiii) Converted
particulate concentrations, where applicable under the department’s rules or
permit.
equation 5-12
|
|
(xiv) Mass
emission rate (lb/hr).
equation 5-15
|
|
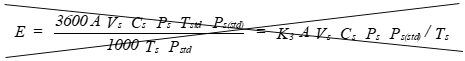
Where:
K3=63.77 for English units.
(xv) Isokinetic variation:
(A) Calculation from raw data.
equation 5-16
|
|
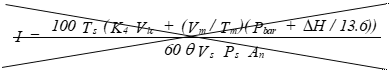
Where:
K4=0.003458 mm Hg - m³ ml - °K for metric units.
=0.002672 in. Hg -
ft.³/ml - °R for English units.
(B) Calculation
from intermediate values.
equation 5-17
|
|
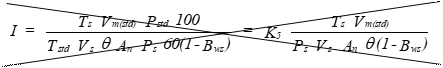
Where:
K5 = 4.307 for metric units.
= 0.09409 for English units.
(xvi) Acceptable results. If
90%=I=110%, the results are acceptable. If the results are low in comparison to
the standard and I is beyond the acceptable range, or if I is less than 90%, the
department may opt to accept the results. Otherwise, reject the results and
repeat the test.
(g) Bibliography:
(i) Federal Register, Volume 42, No. 160, Part 60, Chapter 1, Title 40, Appendix A, Method 5. August 18, 1977.
(ii) Martin, Robert M. Construction Details of Isokinetic Source Sampling Equipment. Environmental Protection Agency. Research Triangle Park, N.C.APTD-0581. April, 1971.
(iii) Rom, Jerome J. Maintenance, Calibration, and Operation of Isokinetic Source Sampling Equipment. Environmental Protection Agency. Research Triangle Park, N.C. APTD-0576. March, 1972.
(iv) Shigehara, R. T. "Adjustments in the EPA Nomograph for Different Pitot Tube Coefficients and Dry Molecular Weights." Stack Sampling News 2:4-11.October, 1974.
(v) Guidelines for Source Testing of Particulate. Michigan Department of Natural Resources, Air Quality Division. June 1, 1977.
R 336.2014 Reference test method 5E.
Rule 1014. Reference method 5E, determination of particulate matter emissions from positive pressure fabric filters, reads as follows:
(a) The principle, applicability, and performance test criteria are as follows:
(i) Principle. Particulate matter is withdrawn isokinetically from the source and collected on a glass fiber filter maintained at a temperature at or above the exhaust gas temperature up to a nominal 248 ±25 degrees Fahrenheit. The particulate mass, which includes any material that condenses at or above the filtration temperature, is determined gravimetrically after the removal of uncombined water.
(ii) Applicability. This method is applicable for the determination of particulate emissions from the stationary sources as identified in table 31 of R 336.1331. The method is also applicable when specifically provided for in the department's rules, orders, a permit to install, or a permit to operate.
(iii) Performance test criteria as follows:
(A) A performance test mustshall consist meet the
requirements under R 336.2003(2).of a minimum of 3 separate samples of a
specific air contaminant conducted within a 36-hour period, unless otherwise
authorized by the department. Each of the 3 separate samples shall be obtained
while the source is operating at a similar production level. For the purpose of
determining compliance with an applicable emission limit, rule, or permit
condition, the arithmetic mean of results of the 3 samples shall apply. If a
sample is accidentally lost or conditions occur in which 1 of the 3 samples
must be discontinued because of forced shutdown, failure of an irreplaceable portion
of the sampling train, extreme meteorological conditions, or other circumstances
beyond the owner's or operator's control, compliance may, upon the approval of
the department, be determined using the arithmetic mean of the results of 2
samples.
(B) For any sources that are is subject
to an emission limitation calculated to 50% excess air, the multipoint, integrated
sampling procedure of R 336.2004(1)(c) shall must be used for gas
analysis. For all other sources that require a determination of the molecular
weight of the exhaust, any an optional sampling procedure of R 336.2004(1)(c)
may be used. Alternatives or modifications to procedures are subject to the approval
of the department.
(C) The minimum volume per sample shall must be 30 cubic feet actual
gas. Minimum sample time shall must be 60 minutes, which may be continuous
or a combination of shorter sampling periods for sources that operate in a cyclic
manner. Smaller sampling times or sample volumes, when necessitated by process variables
or other factors, may be approved by the department.
(D) For any a source whose
emission control device alters the moisture content of the exhaust gas, a moisture
determination shall must be performed in a location upstream from the
emission control device and in accordance with R 336.2004(1)(d) or an
alternative method approved by the department.
(b) The following provisions apply to apparatus:
(i) Sampling train. A schematic of the
sampling train used in this method is shown in figure 103 under R 336.2021.
Construction details for many, but not all, of the train components are given
in APTD-0581, (subdivision
(g)(ii) of this rule). For changes from the APTD-0581 document and for allowable
modifications to figure 103 under
R 336.2021, consult with the department. The operating and maintenance
procedures for many, but not all, of the sampling train are described in APTD-0576, adopted by
reference in R 336.1902 and as referenced under (subdivision
(g)(iii) of this rule). Since correct usage is important in obtaining
valid results, all users shall read APTD-0576 and adopt the applicable operating
and maintenance procedures outlined in it, unless otherwise specified in these rules.
The sampling train consists of the following components:
(A) Probe nozzle. Stainless steel (316)
or glass with sharp, tapered leading edge. The angle of taper shall must be less than 30
degrees and the taper shall must be on the outside to preserve a constant
internal diameter. The probe nozzle shall must be of the button-hook
design, unless otherwise specified by the department. If made of stainless
steel, the nozzle shall must be constructed from seamless
tubing. Other materials of construction may be used, subject to the approval of
the department. A range of nozzle sizes suitable for isokinetic sampling shall must be available, for
example, 0.32 to 1.27
centimeters, (1/8 to
1/2
in.ch,) or larger if higher
volume sampling trains are used inside diameter (ID) nozzles in
increments of 0.16
centimeter, (1/16 in.ch). Each nozzle shall must be calibrated
according to the procedures outlined in subdivision (e) of this rule.
(B) Probe liner. Borosilicate or
quartz glass tubing with a heating system capable of maintaining a gas
temperature at the exit end during sampling of 120 ±14 degrees Centigrade, (248 ±25
degrees Fahrenheit), another temperature as specified by the department's
rules, or a temperature approved by the department for a particular
application. The tester may opt to operate the equipment at a temperature lower
than that specified. Since the actual temperature at the outlet of the probe is
not usually monitored during sampling, probes constructed according to
APTD-0581 which that utilize the calibration curves of APTD-0576,
or calibrated according to the procedure outlined in APTD-0576, are acceptable.
Either borosilicate or quartz glass probe liners may be used for stack
temperatures up to about 480 degrees Centigrade, (900 degrees
Fahrenheit);. quartz Quartz liners shall must be used for
temperatures between 480 and 900 degrees Centigrade, (900 and 1,650
degrees Fahrenheit). Both types of liners may be used at higher
temperatures than specified for short periods of time, subject to the approval
of the department. The softening temperature for borosilicate is 820 degrees
Centigrade, (1,508
degrees Fahrenheit,) and for quartz it
is 1,500 degrees Centigrade, (2,732 degrees Fahrenheit).When
practical, every effort shall must be made to use borosilicate or quartz
glass probe liners. Alternatively, metal liners, such as 316 stainless steel,
Incoloy 825, or other corrosion-resistant materials made of seamless tubing,
may be used, subject to the approval of the department.
(C) Pitot tube. Type S, as described
in section 2.1 of method 2, or other device approved by the department.
The pitot tube shall must be attached to the probe, as shown in
figure 103 under R 336.2021, to allow constant monitoring of the stack gas
velocity. The impact, (high
pressure,) opening plane of the
pitot tube shall must be even with or above the nozzle entry
plane, (see
method 2, figure 2-6b Velocity Traverse Data) during sampling.
The type S pitot tube assembly shall must have a known
coefficient, determined as outlined in section 4 of method 2.
(D) Differential pressure gauge.
Incline manometer or equivalent devices, (quantity 2), as
described in section 2.2 of method 2. One manometer shall must be used for
velocity head (p) readings, and the other shall must be used for
orifice differential pressure readings.
(E) Filter holders. Two separate
filter holders in series or 1 filter holder with separate filter supports and
seals for 2 filters. One filter holder with 2 filters held in contact with each
other is not acceptable. Materials of construction may be stainless steel (316),
glass, teflonTeflon, or such other
material approved by the department.
(F) Filter heating system. Any heating
system capable of maintaining a temperature around the filter holder during sampling
of 120 ±14 degrees Centigrade, (248 ±25 degrees Fahrenheit),
another temperature as specified by the department's rules or a permit
condition, or a temperature approved by the department for a particular application.
Alternatively, the tester may opt to operate the equipment at a temperature lower
than that specified. A temperature gauge capable of measuring temperature to
within 3 degrees Centigrade, (5.4 degrees Fahrenheit), shall must be installed so that
the temperature around the filter holders can be regulated and monitored during
sampling. Heating systems other than the one shown in APTD-0581 may be used.
(G) Condenser. The following system shall must be used to
determine the stack gas moisture content: Three impingers connected in series with
leak-free ground glass fittings or any similar leak-free noncontaminating
fittings. All impingers shall must be of the Greenburg-Smith design and
shall
must
be modified by replacing the tip with a 1.3 centimeters, (1/2 in.ch,) ID inside diameter glass tube
extending to about 1.3
centimeters, (1/2 in.ch,) from the bottom of
the flask. Modifications, such as using flexible connections between the
impingers or using materials other than glass, are permitted allowed subject to the approval
of the department. The first impinger shall must contain a known quantity
of water, (as described in subdivision (d)(i)(C)
of this rule), the second shall must be empty, and the
third shall
must
contain a known weight of silica gel or equivalent desiccant. Alternatively,
any system that cools the sample gas stream and allows measurement of the water
condensed and moisture leaving the condenser, each to within 1 milliliter or 1 gram, may be used
subject to the approval of the department. In any case, the means for measuring
the moisture leaving the condenser shall must be by passing the
sample gas stream through a tared silica gel, or equivalent desiccant, trap
with exit gases kept below 20 degrees Centigrade, (68
degrees Fahrenheit,) and determining
the weight gain. If a determination of the particulate matter collected in the impingers
is required by the department's rules, a permit to install, or a permit to
operate, the impinger system described in this subdivision shall must be used, without
modification. Contact the department as to the sample recovery and analysis of
the impinger contents.
(H) Metering system. Vacuum gauge,
leak-free pump, thermometers capable of measuring temperature to within 3 degrees
Centigrade, (5.4
degrees Fahrenheit), dry-gas meter capable of measuring volume to within
2%, and related equipment as shown in figure 103 under R 336.2021. Other
metering systems capable of maintaining sampling rates within 10% of isokinetic
and capable of determining sample volumes to within 2% may be used, subject to
the approval of the department. When the metering system is used in conjunction
with a pitot tube, the system shall must enable checks of
isokinetic rates. Sampling trains utilizing metering systems designed for higher
flow rates than those described in APTD-0581 or APTD-0576, adopted by
reference in R 336.1902, may be used if the specifications of this rule are
met.
(I) Barometer. Mercury, aneroid, or other
barometer capable of measuring atmospheric pressure to within 2.5 millimeters Hgmercury, (0.1-inch. Hgmercury). In many cases, the
barometric reading may be obtained from a nearby national weather service
station. When obtained from this source, the station value, which is the
absolute barometric pressure, shall must be requested and an
adjustment for elevation differences between the weather station and sampling
point shall
must
be applied at a rate of minus 2.5 millimeters Hgmercury, (0.1-in.ch Hgmercury,) per 30 meters, (100 foot,.) elevation
increase or vice versa for elevation decrease.
(J) Gas density determination equipment.
Temperature sensor and pressure gauge, as described in sections 2.3 and 2.4
of method 2, and gas analyzer, if necessary, as described in method 3. The temperature
sensor shall
must,
preferably, be permanently attached to the pitot tube or sampling probe in a
fixed configuration such so that the tip of the sensor extends beyond
the leading edge of the probe sheath and does not touch any metal.
Alternatively, the sensor may be attached just before use in the field. Note,
however, that if the temperature sensor is attached in the field, the sensor shall must be placed in an interference-free
arrangement with respect to the type S pitot tube openings, (see
method 2, figure 2.76 Velocity Traverse Data).As a second alternative,
if a difference of not more than 1% in the average velocity measurement is to be
introduced, the temperature gauge need not be attached to the probe or pitot
tube. This alternative is subject to the approval of the department.
(ii) Sample recovery. The following items are needed:
(A) Probe-liner and probe-nozzle brushes.
Nylon bristle brushes with stainless steel wire handles. The probe brush shall must have extensions, at
least as long as the probe, made of stainless steel, nylon, teflonTeflon, or similarly
inert material. The brushes shall must be properly sized and shaped to
brush out the probe liner and nozzle.
(B) Wash bottles--2bottles - 2. Glass wash
bottles are recommended.; polyethylene Polyethylene wash bottles may
be used at the option of the tester. It is recommended that acetone not be
stored in polyethylene bottles for longer than a month.
(C) Glass sample storage containers. Chemically
resistant, borosilicate glass bottles, for acetone washes, 500 milliliters or 1000 milliliters. Screw cap liners
shall
must
either be rubber-backed teflonTeflon or shall must be constructed so
as to be leak-free and resistant to chemical attack by acetone. Narrow-mouth glass
bottles have been found to be less prone to leakage. Alternatively,
polyethylene bottles may be used.
(D) Filter containers. Glass, polyethylene, or aluminum tube containers, unless otherwise specified by the department.
(E) Graduated cylinder or balance. To
measure condensed water to within 1 milliliter or 1 gram. Graduated
cylinders shall
must
have subdivisions of not more than 2 milliliters. Most laboratory
balances are capable of weighing to the nearest 0.5 gram or less. Any of
these balances are suitable for use here and in paragraph (iii)(D) of this
subdivision.
(F) Plastic storage containers. Airtight containers to store silica gel.
(G) Funnel and rubber policeman., Tto aid in the transfer
of silica gel to container;, but not necessary if silica gel is
weighed in the field.
(H) Funnel. Glass or polyethylene, to aid in sample recovery.
(iii) Analysis. The following equipment is needed for analysis:
(A) Glass weighing dishes.
(B) Desiccator.
(C) Analytical balance. To measure to within 0.1 milligrams.
(D) Balance. To measure to within 0.5 milligrams.
(E) Beakers. 250 milliliters.
(F) Hygrometer., Tto measure the relative
humidity of the laboratory environment.
(G) Temperature gauge., Tto measure the temperature
of the laboratory environment.
(c) The following provisions apply to reagents:
(i) Sampling. The reagents used in sampling are as follows:
(A) Filters. Two outstack filters may
be any combination of alundum ceramic thimble filters, type RA-98 or glass
fiber filters, type A without organic binder. The size of such filters shall must allow proper sampling
rates to maintain isokinetics using the nozzle sizes specified in subdivision (b)(i)(A)
of this rule. Alternatively, other types of filters may be used, subject to the
approval of the department.
(B) Silica gel. Indicating type, 6 to
16 mesh. If previously used, dry at 175 degrees Centigrade, (350
degrees Fahrenheit,) for 2 hours. New
silica gel may be used as received. Alternatively, other types of desiccants,
the equivalent or better of silica gel, may be used, subject to the approval of
the department.
(C) Water. When analysis of the material
caught in the impingers is required, distilled water shall must be used. Run
blanks before field use to eliminate a high blank on test samples.
(D) Crushed ice.
(E) Stopcock grease.
Acetone-insoluble, heat-stable silicone grease. This is not necessary if
screw-on connectors with teflonTeflon sleeves, or equivalent, are used.
Alternatively, other types of stopcock grease may be used, subject to the
approval of the department.
(ii) Sample recovery. Washing solvent.
Either acetone or distilled water may be used for sample recovery. If acetone
is used for washing solvent, then reagent grade, less than 0.001% residue, in glass
bottles is required. Acetone from metal containers generally has a high residue
blank and shall
must
not be used. Suppliers sometimes transfer acetone to glass bottles from metal
containers.; thus Thus, acetone blanks shall must be run before field
use, and only acetone with low blank values, less than 0.001%, shall must be used. In no case
shall
must
a blank value of more than 0.001% of the weight of acetone used be subtracted
from the sample weight. If distilled water is used for washing solvent, use distilled
water with less than 0.001% residue. Run blanks before field use to eliminate a
high blank on test samples.
(iii) Analysis. Two reagents are required for the analysis:
(A) Solvent. Same as paragraph (ii) of this subdivision for quantitative transfer.
(B) Desiccant. Anhydrous calcium sulfate, indicating type. Alternatively, other types of desiccants may be used, subject to the approval of the department.
(d) The following provisions apply to procedures:
(i) Determination of single
measurement sites. The measurement site for a positive pressure fabric filter with
an exhaust stack meeting method 1 criteria shallmust be in accordance with
section 2.1 of method 1. The measurement site for positive pressure
fabric filters with short stacks or physical configuration not amenable to the
requirements of method 1 shall must be determined from the following alternatives,
or as approved by the department:
(A) Short stacks not meeting method 1
criteria: Short stacks may be extended in accordance with the procedures set
forth in method 1 or by the use of flow straightening vanes. The flow
straightening vanes shall must be of the egg crate design, (see
figure 109 under R 336.2021). The measurement site, when using
straightening vanes, shall must be at a distance not less than 2 times
the average equivalent diameter of the vane opening and not less than 1/2half of the overall stack
diameter upstream of the stack outlet.
(B) Roof monitor or monovent exhaust outlets:
For positive pressure fabric filters equipped with peaked roof monitors, ridge
vents, or other types of monovents, use a measurement site at the base of the monovent.
Examples of such the locations are shown in figure 108 under
R 336.2021. The measurement site shall must be upstream of
any exhaust point.
(C) Measurement site in fabric filter compartment housing. Sample immediately downstream of the filter bags directly as shown in the examples in figure 108 under R 336.2021. Depending on the housing design, use sampling ports in the housing walls or locate the sampling equipment within the compartment housing.
(ii) Determination of number and
location of traverse points. The number and location of traverse points for
single exhaust stacks on positive pressure fabric filters meeting method 1
criteria shallmust be in accordance
with section 2.3 of method 1. The number of traverse points for other single
measurement sites not meeting method 1 criteria shall must not be less than 24.
For example, a rectangular measurement site, such as a monovent, would require
the use of a balanced 5-by-5 traverse point matrix. All traverse points shall must be sampled for
each test run.
(iii) Multiple measurement sites. Sampling from 2 or more stacks or measurement sites may be combined for a test run, if all of the following requirements are met:
(A) All measurement sites up to 12 shall must be sampled. For more
than 12 measurement sites, conduct sampling on not less than 12 sites or 50% of
the sites, whichever is greater. The measurement sites sampled shall must be evenly, or
nearly evenly, distributed among the available sites, if not all of the sites
are to be sampled.
(B) The same number of measurement
sites shall
must
be sampled for each test run.
(C) The minimum number of traverse points per test run is 24. An exception to the 24-point minimum would be a test combining the sampling from 2 stacks meeting method 1 criteria for acceptable stack length, and method 1 specifies fewer than 12 points per site.
(D) As long as the 24 traverse points
per test run criterion is met, the number of traverse points per measurement
site may be reduced to 8. Alternatively, conduct a test run for each measurement
site individually using the criteria in this paragraph and paragraph (ii) of this
subdivision for the number of
traverse points. Each test shall must count toward the total of 3
required for a performance test. If more than 3 measurement sites are sampled,
the number of traverse points per measurement site may be reduced to 8 if not
less than 72 traverse points are sampled for all 3 tests.
(iv) Sampling. The complexity of this
method is such that, in order to obtain reliable results, testers shall must be trained and experienced
with the test procedures. Sampling shall must comply with the
following provisions:
(A) Pretest preparation. All the components
shall
must
be maintained and calibrated according to the applicable procedures described in
APTD-0576,
adopted by reference in R 336.1902, unless otherwise specified in this rule.
Weigh several 200 to 300 gram portions of silica gel in airtight containers
to the nearest 0.5
gram. Record the total
weight of the silica gel plus container on each container. As an alternative,
the silica gel need not be preweighed, but may be weighed directly in its
impinger or sampling holder just before train assembly. Check filters visually against
light for irregularities, flaws, pinhole leaks, or cracks. Label filters of the
proper size on the back side using numbering machine ink. As an alternative, label
the shipping containers, (subdivision (b)(ii)(D) of this rule,) and keep the filters
in these containers at all times, except during sampling and weighing. Dry the filters
in an oven at 105 degrees Centigrade, (220 degrees Fahrenheit,) for a minimum of
2 hours, cool for at least 1 hour in a desiccator containing anhydrous
calciumsulfate, and individually weigh and record their weights to the nearest
0.1
milligram. During the
weighing, the filters shall must not be exposed to the laboratory
atmosphere for a period of more than 2 minutes and a relative humidity above 50%.
Procedures, other than those specified, that account for relative humidity
effects may be used, subject to the approval of the department.
(B) Preliminary determinations.
Select the sampling site and the minimum number of sampling points according to
method 1 or as specified by the department. Determine the stack pressure, temperature,
and the range of velocity heads using method 2.; it It is recommended that
a leak check of the pitot lines, (see method 2, section 3.1), be performed.
Determine the moisture content using approximation method 4, or its
alternatives, for the purpose of making isokinetic sampling rate settings. Determine
the stack gas dry molecular weight, as described in method 2, section 3.6; if. If integrated method
3 sampling is used for molecular weight determination, the integrated bag sample
shall must be taken
simultaneously with, and for the same total length of time as, the particulate
sample run. Select a nozzle size based on the range of velocity heads so that
it is not necessary to change the nozzle size in order to maintain isokinetic sampling
rates. During the run, do not change the nozzle size. Ensure that the proper
differential pressure gauge is chosen for the range of velocity heads
encountered, (see section
2.2 of method 2). Select a suitable probe liner and probe length so
that all traverse points can be sampled. For large stacks, consider sampling
from opposite sides of the stack to reduce the length of probes. Select a total
sampling time greater than or equal to the minimum total sampling time
specified in the test procedures for the specific industry so that the sampling
time per point is not less than 5 minutes, unless approved by the department, or
some greater time interval as specified by the department, and so that the sample
volume taken, corrected to standard conditions, exceeds the required minimum
total gas sample volume. The latter is based on an approximate average sampling
rate. It is recommended that the number of minutes sampled at each point be an
integer or an integer plus 1/2 minute to avoid timekeeping errors. In some
circumstances, such as in batch cycles, it may be necessary to sample for
shorter times at the traverse points and to obtain smaller gas sample volumes.
In these cases, the department's approval shall must first be
obtained.
(C) Preparation of collection train.
During preparation and assembly of the sampling train, keep all openings where
contamination can occur covered until just before assembly or until sampling is
about to begin. Place 100 milliliters of water in the first impinger,
leave the second impinger empty, and transfer approximatlely 200 to 300 grams of preweighed silica
gel from its container to the third impinger. More silica gel may be used, but
care should be taken to ensure that it is not entrained and carried out from
the impinger during sampling. Place the container in a clean place for later use
in the sample recovery. Alternatively, the weight of the silica gel plus impinger
may be determined to the nearest 0.5 gram and recorded. Using tweezers or clean disposable
surgical gloves, place a labeled, (identified,) and weighed
filter in the filter holder. Be sure that the filter is properly centered and
the gasket properly placed so as to prevent the sample gas stream from
circumventing the filter. Check the filter for tears after assembly is
completed. When glass liners are used, install the selected nozzle using a Viton
A O0-ring when stack
temperatures are less than 260 degrees Centigrade, (500 degrees
Fahrenheit,) and an asbestos string
gasket when temperatures are higher. See APTD-0576, adopted by
reference in R 336.1902, for details. Other connecting systems using either 310
stainless steel or teflon Teflon ferrules may be used. When metal
liners are used, install the nozzle in the same manner as for glass liners or
by a leak-free direct mechanical connection. Mark the probe with heat-resistant
tape or by some other method to denote the proper distance into the stack or duct
for each sampling point. Set up the train as in figure 103 under R 336.2021.
If necessary, use a very light coat of silicone grease on all ground glass
joints. Grease only the outer portion, (see APTD-0576,) to avoid the
possibility of contamination by the silicone grease. Place crushed ice around
the impingers.
(D) Leak check procedures:
(I1) Pretest leak
check. A pretest leak check is strongly recommended, but not required, to
prevent invalid sampling and wasted time. If the tester opts to conduct the
pretest leak check, the following procedure shall must be used: After
the sampling train has been assembled, turn it on and set the filter and probe
heating systems at the desired operating temperatures. Allow time for the
temperatures to stabilize. If a Viton A O0-ring or other leak-free connection
is used in assembling the probe nozzle to the probe liner, leak check the train
at the sampling site by plugging the nozzle and pulling a 380 millimeter Hgmercury, (15 in.ch Hgmercury,) vacuum. A lower
vacuum may be used if it is not exceeded during the test. If an asbestos string
is used, do not connect the probe to the train during the leak check. Instead,
leak check the train by first plugging the inlet to the filter holder, (cyclone,
if applicable,) and pulling a 380 millimeter Hgmercury, (15 in.ch Hgmercury,) vacuum. A lower
vacuum may be used if it is not exceeded during the test. Then connect the
probe to the train and leak check at about a 25 millimeter Hgmercury, (1 in.ch Hgmercury,) vacuum.; alternatively
Alternatively, the probe may be
leak checked with the rest of the sampling train, in 1 step, at a 380 millimeter Hgmercury, (15 in.ch Hgmercury,) vacuum. Leakage
rates in excess of 4% of the average sampling rate or 0.00057 cubic meters per
minute, m³/min
(0.02
cubic
feet per minute,cfm), whichever is
less, are unacceptable. The following leak check instructions for the sampling train
described in APTD-0576 and APTD-058 may be helpful. Start the pump with the
bypass valve fully open and the coarse adjust valve completely closed.
Partially open the coarse adjust valve and slowly close the bypass valve until the
desired vacuum is reached. Do not reverse the direction of the bypass valve, as; this will cause
water to back up into the filter holder. If the desired vacuum is exceeded,
either leak check at this higher vacuum or end the leak check and start over.
When the leak check is completed, first slowly remove the plug from the inlet
to the probe, filter holder, or cyclone, if applicable, and immediately turn
off the vacuum pump. This prevents the water in the impingers from being forced
backward into the filter holder and prevents silica gel from being entrained
backward into the third impinger.
(II2) Leak checks during
sample run. If, during the sampling run, a component, such as a filter assembly
or impinger, change becomes necessary, a leak check shall must be conducted
immediately before the change is made. The leak check shall must be done according
to the procedure outlined in paragraph (iv)(D)(I1) of this
subdivision, except that it shall must be done at a vacuum equal to or
greater than the maximum value recorded up to that point in the test. If the
leakage rate is found to be not more than 0.00057 cubic meters per
minute, m³/min
(0.02
cubic
feet per minute, cfm)
or
4% of the average sampling rate, whichever is less, the results are acceptable and
no correction need be applied to the total volume of dry gas metered.; if If, however, a higher
leakage rate is obtained, the tester shall either record the leakage rate and plan
to correct the sample volume, as shown in subdivision (f)(iii) of R 336.2011this rule, or shall void
the sampling run. Immediately after component changes, leak checks are optional.; if such If the leak checks are
done, the procedure outlined in paragraph (iv)(D)(I1) of this
subdivision shall must be used.
(III3) Post-test leak
check. A leak check is mandatory at the conclusion of each sampling run. The
leak check shall must be done in accordance with the procedures
outlined in paragraph (iv)(D)(I1) of this subdivision, except that
it shall must be conducted at a
vacuum equal to or greater than the maximum value reached during the sampling
run. If the leakage rate is found to be not more than 0.00057 cubic meters per
minute, m³/min
(0.02
cubic
feet per minute,cfm) or 4% of the average
sampling rate, whichever is less, the results are acceptable and no correction need
be applied to the total volume of dry gas metered. If, however, a higher leakage
rate is obtained, the tester shall either record the leakage rate and correct the
sample volume, as shown in subdivision (f)(iii) of R 336.2011this rule, or shall void
the sampling run.
(E) Particulate train operation. During
the sampling run, maintain an isokinetic sampling rate that is within 10% of true
isokinetic, unless otherwise specified by the department. For each run, record
the data required on a data sheet such as the one shown in figure 104 under
R 336.2021. Be sure to record the initial dry-gas meter reading. Record the
dry-gas meter readings at the beginning and end of each sampling time
increment, when changes in flow rates are made, before and after each leak
check, and when sampling is halted. Take other readings required by figure 104
under R 336.2021 at least once at each sample point during each time
increment, and take additional readings when significant changes, 20% variation
in velocity head readings, necessitate additional adjustments in flow rate.
Level and zero the manometer. Because the manometer level and zero may drift
due to vibrations and temperature changes, make periodic checks during the
traverse. Clean the portholes before the test run to minimize the chance of sampling
deposited material. To begin sampling, remove the nozzle cap and verify that
the pitot tube and probe are properly positioned. Position the nozzle at the
first traverse point with the tip pointing directly into the gas stream.
Immediately start the pump and adjust the flow to isokinetic conditions.
Nomographs that aid in the rapid adjustment of the isokinetic sampling rate
without excessive computations are available. These nomographs are designed for
use when the type S pitot tube coefficient is 0.85 ±0.02 and the stack gas
equivalent density, (dry
molecular weight,) is equal to 29 ±4.
APTD-0576,
adopted by reference in R 336.1902, details the procedure for using the
nomographs. If Cp and Md are outside the above stated ranges, do not use the
nomographs unless appropriate steps, (see subdivision (g)(iv) of this
rule,) are taken to
compensate for the deviations. When the stack is under significant negative pressure, (height of
impinger stem), take care to pull low flow when inserting the probe into
the stack to prevent water from backing into the sample tubing and to avoid pulsation
through the filter and possible loss of materials. When the probe is in
position, block off the openings around the probe and porthole to prevent
unrepresentative dilution of the gas stream. Traverse the stack cross section,
as required by method 1 or as specified by the department, being careful not to
bump the probe nozzle into the stack walls when sampling near the walls or when
removing or inserting the probe through the portholes.; this This minimizes the chance
of extracting deposited material. During the test run, add more ice and, if necessary,
salt to maintain a temperature of less than 20 degrees Centigrade, (68
degrees Fahrenheit,) at the
condenser/silica gel outlet. Also, periodically check the level and zero of the
manometer. If the pressure drop across the filter becomes too high and makes
isokinetic sampling difficult to maintain, the filter may be replaced in the midst
of a sample run. It is recommended that another complete filter assembly be used
rather than attempting to change the filter itself. Before a new filter
assembly is installed, conduct a leak check, (see paragraph (iv)(D)(2II) of this subdivision).
The total particulate weight shall must include the
summation of all filter assembly catches. A single train shall must be used for the
entire sample run, except in cases where simultaneous sampling is required in 2
or more separate ducts, at 2 or more different locations within the same duct, or
where equipment failure necessitates a change of trains. In all other
situations, the use of 2 or more trains shall must be subject to the
approval of the department. Note that when 2 or more trains are used, separate
analyses of the front-half catches from the individual trains may be combined, as
may the impinger catches, and 1 analysis of the front-half catch and 1 analysis
of impinger catch may be performed. Consult with the department for details concerning
the calculation of results when 2 or more trains are used. At the end of the
sample run, turn off the coarse adjust valve, remove the probe and nozzle from
the stack, turn off the pump, record the final dry-gas meter reading, and
conduct a post-test leak check, as outlined in paragraph (iv)(D)(III3) of this subdivision.
Also, leak-check the pitot lines as described in method 2, section 3.1; the
The
lines
shall must pass this leak
check to validate the velocity head data.
(F) Calculation of percent isokinetic.
Calculate percent isokinetic, (see subdivision (f) of this rule,) to determine whether
the run was valid or whether another test run should be made. If there was
difficulty in maintaining isokinetic rates due to source conditions, consult with
the department for possible variance on the isokinetic rates.
(v) Sample recovery. Proper cleanup
procedure begins as soon as the probe is removed from the stack at the end of
the sampling period. Allow the probe to cool. When the probe can be safely handled,
wipe off all external particulate matter near the tip of the probe nozzle and
place a cap over it to prevent losing or gaining particulate matter. Do not cap
off the probe tip tightly while the sampling train is cooling down as this creates
a vacuum in the filter holder and draws water from the impingers into the
filter holder. Before moving the sample train to the cleanup site, remove the
probe from the sample train, wipe off the silicone grease, and cap the open outlet
of the probe. Be careful not to lose any condensate that might be present. Wipe
off the silicone grease from the filter inlet where the probe was fastened and
cap it. Remove the umbilical cord from the last impinger and cap the impinger.
If a flexible line is used between the first impinger or condenser and the
filter holder, disconnect the line at the filter holder and let any condensed
water or liquid drain into the impingers or condenser. After wiping off the
silicone grease, cap off the filter holder outlet and impinger inlet.
Ground-glass stoppers, plastic caps, or serum caps may be used to close these
openings. Transfer the probe and filter-impinger assembly to the cleanup area. This
area shall must be clean and
protected from the wind so that the chances of contaminating or losing the
sample are minimized. Save a portion of the solvent used for cleanup as a
blank. Take 200
milliliters of this solvent directly
from the wash bottle being used and place it in a glass sample container
labeled "solvent blank." Inspect the train before and during disassembly
and note any abnormal conditions. Treat the samples as follows: Container Nos.numbers 1, 1A. Carefully
remove the filters from the filter holders and place in their identified
containers. Use a pair of tweezers or clean disposable surgical gloves, or both,
to handle the filters. Carefully transfer to the container any particulate
matter or filter fibers, or both, that adhere to the filter holder gasket by
using a dry nylon bristle brush or sharp-edged blade, or both. Seal the
container. Container No.number 2. Taking care to see that dust on
the outside of the probe or other exterior surfaces does not get into the sample,
the testoer shall
quantitatively recover from particulate matter or any condensate from the
nozzle, probe fitting, probe liner, and from both filter holders by washing
these components with solvent and placing the wash in a glass container. Perform
the solvent rinses as follows: Carefully remove the probe nozzle and clean the inside
surface by rinsing with solvent from a wash bottle and brushing with a nylon bristle
brush. Brush until the solvent rinse shows no visible particles and then make a
final rinse of the inside surface with solvent. Brush and rinse the inside
parts of the Swagelok fitting with solvent in a similar way until no visible
particles remain. Rinse the probe liner with solvent by tilting and rotating the
probe while squirting solvent into its upper end so that all inside surfaces are
wetted with acetone. Let the solvent drain from the lower end into the sample
container. A glass or polyethylene funnel may be used to aid in transferring
liquid washes to the container. Follow the solvent rinse with a probe brush.
Hold the probe in an inclined position and squirt solvent into the upper end as
the probe brush is being pushed with a twisting action through the probe.; hold Hold a sample
container underneath the lower end of the probe and catch any solvent and
particulate matter that is brushed from the probe. Run the brush through the
probe 3 or more times until no visible particulate matter is carried out with
the solvent or until none remains in the probe liner on visual inspection. With
stainless steel or other metal probes, run the brush through, in the manner set
forth in this paragraph, not less than 6 times, since metal probes have small
crevices in which particulate matter can be entrapped. Rinse the brush with solvent
and quantitatively collect these washings in the sample container. After the
brushing, make a final solvent rinse of the probe as described above. It is
recommended that 2 people be used to clean the probe to minimize sample losses.
Between sampling runs, keep brushes clean and protected from contamination.
After ensuring that all joints have been wiped clean of silicone grease, clean
the inside of both filter holders by rubbing the surfaces with a nylon bristle
brush and rinsing with solvent. Rinse each surface 3 times, or more if needed,
to remove visible particulate. Make a final rinse of the brush and filter
holder. After all solvent washings and particulate matter have been collected
in the sample container, tighten the lid on the sample container so that solvent
will not leak out when it is shipped to the laboratory. Mark the height of the fluid
level to determine whether or not leakage occurred during transport. Label the
container to clearly identify its contents. Container No.number 3. Note the color
of the indicating silica gel to determine if it has been completely spent and
make a notation of its condition. Transfer the silica gel from the third
impinger to its original container and seal. A funnel may make it easier to
pour the silica gel without spilling it. A rubber policeman may be used as an
aid in removing the silica gel from the impinger. It is not necessary to remove
the small amount of dust particles that adhere to the impinger wall and are
difficult to remove. Since the gain in weight is to be used for moisture
calculations, do not use any water or other liquids to transfer the silica gel.
If a balance is available in the field, follow the procedure for container No.number 3 in paragraph (vi)
of this subdivision. Impinger water. Treat the impingers as follows: Make a
notation of any color or film in the liquid catch. Measure the liquid that is in
the first 2 impingers to within ±1 milliliter by using a
graduated cylinder or by weighing it to within ±1.0 gram by using a
balance if none is available. Record the volume or weight of liquid present.
This information is required to calculate the moisture content of the effluent
gas. Discard the liquid after measuring and recording the volume or weight,
unless analysis of the impinger catch is required, (see
subdivision (b)(i)(G) of this rule). If a different type of condenser is
used, measure the amount of moisture condensed either volumetrically or
gravimetrically. When possible, containers shall must be shipped in a manner
that keeps them upright at all times.
(vi) Analysis. Record the data
required on a sheet such as the one shown in figure 106 under R 336.2021. Handle each
sample container as follows: Container Nos.numbers 1, 1A. Analyze
and report each filter separately. Transfer the filter and any loose
particulate from the sample container to a tared-glass weighing dish. Dry the
filter in an oven at 105 degrees Centigrade, (220 degrees Fahrenheit,) for a minimum of
2 hours, cool for at least 1 hour in a desiccator containing anhydrous calcium
sulfate, and weigh and record its weight to the nearest 0.1 milligram. During the
weighing, the filter shall must not be exposed to the laboratory
atmosphere for a period of more than 2 minutes or a relative humidity above
50%. Procedures, other than those specified, that account for relative humidity
effects may be used, subject to the approval of the department. The method used
for the drying and weighing of filters shall must be consistent
before and after the test. Container No.number 2. Note the level
of liquid in the container and confirm on the analysis sheet whether or not leakage
occurred during transport. If a noticeable amount of leakage has occurred,
either void the sample or use methods, subject to the approval of the department,
to correct the final results. Measure the liquid in this container either volumetrically
to ±1
milliliters or
gravimetrically to ±1.0 grams. Transfer the contents to a tared 250-
milliliter beaker and
evaporate to dryness either at ambient temperature and pressure for acetone or
at 95 degrees Centigrade, (203 degrees Fahrenheit,) in an oven for
distilled water. Then subject the sample to 250 degrees Centigrade, (482 degrees Fahrenheit,) in an oven for 2
to 3 hours. Desiccate for 24 hours and weigh to a constant weight. Report the
results to the nearest 0.1 milligram. Container No.number 3. Weigh the
spent silica gel, or silica gel plus impinger, to the nearest 0.5 gram using a balance.
This step may be conducted in the field. "Solvent blank" container. Measure
solvent in this container either volumetrically or gravimetrically. Transfer
the contents to a tared 250- milliliter beaker and
evaporate to dryness either at ambient temperature and pressure for acetone or
at 95 degrees Centigrade, (203 degrees Fahrenheit,) in an oven for
distilled water. Then subject the sample to 250 degrees Centigrade, (482
degrees Fahrenheit,) in an oven for 2
to 3 hours. Desiccate for 24 hours and weigh to a constant weight. Report the
results to the nearest 0.1 milligram. If acetone is used, the contents of
container No.number 2, as well as the
acetone blank container, may be evaporated at temperatures higher than ambient.
If evaporation is done at an elevated temperature, the temperature shall
must be closely supervised,
and the contents of the beaker shall must be swirled
occasionally to maintain an even temperature. Use extreme care, as acetone is
highly flammable and has a low flash point.
(e) Calibration. Maintain a laboratory log of all calibrations. The following provisions apply to calibrations:
(i) Probe nozzle. Probe nozzles shall
must be calibrated
before their initial use in the field. Using a micrometer, measure the inside
diameter of the nozzle to the nearest 0.025 millimeter, (0.001 in.ch). Make 3 separate measurements
using different diameters each time and obtain the average of the measurements.
The difference between the high and low numbers shall must not exceed 0.1 millimeter, (0.004 in.ch). When nozzles
become nicked, dented, or corroded, they shall the nozzles must be reshaped,
sharpened, and recalibrated before use. Each nozzle shall must be permanently
and uniquely identified.
(ii) Pitot tube. The type S pitot tube
assembly shall must be calibrated
according to the procedure outlined in section 4 of method 2.
(iii) Metering system. Before its initial
use in the field, the metering system shallmust be calibrated
according to the procedure outlined in APTD-0576, adopted by
reference in R 336.1902. Instead of physically adjusting the dry-gas meter dial
readings to correspond to the wet-test meter readings, calibration factors may be
used to mathematically correct the gas meter dial readings to the proper values.
Before calibrating the metering system, it is suggested that a leak check be
conducted. For metering systems having diaphragm or rotary pumps, the normal
leak check procedure will not detect leakages within the pump. For these cases,
the following leak check procedure is suggested: Make a 10-minute calibration
run at 0.00057 cubic
meters per minute, m³/min (0.02 cubic feet per minute,cfm); at the end of the
run, take the difference of the measured wet-test meter and dry-gas meter volumes, and; divide the
difference by 10 to get the leak rate. The leak rate shall must not exceed
0.00057 cubic
meters per minute, m³/min (0.02 cubic feet per minute,cfm).After each field
use, the calibration of the metering system shall must be checked by
performing 3 calibration runs at a single, intermediate orifice setting, based
on the previous field test, with the vacuum set at the maximum value reached
during the test series. To adjust the vacuum, insert a valve between the
wet-test meter and the inlet of the metering system. Calculate the average
value of the calibration factor. If the calibration has changed by more than
5%, recalibrate the meter over the full range of orifice settings, as outlined
in APTD-0576. Alternatively, a spirometer may be substituted for a wet-test meter
in the above mentioned calibration procedures. Alternative procedures, such as
using the orifice meter coefficients, may be used, subject to the approval of
the department. If the dry-gas meter coefficient values obtained before and after
a test series differ by more than 5%, the test series shall must be performed using
whichever meter coefficient value, before or after, gives the lower value of
total sample volume.
(iv) Probe heater calibration. The probe
heating system shall must be calibrated before its initial use in
the field according to the procedures outlined in APTD-0576, adopted by
reference in R 336.1902. Probes constructed according to APTD-0581 need not
be calibrated if the calibration curves in APTD-0576 are used.
(v) Temperature gauges. Use the
procedure in section 4.3 of method 2 to calibrate instack temperature
gauges. Dial thermometers, such as those used for the dry-gas meter and condenser
outlet, shall must be calibrated
against mercury-in-glass thermometers.
(vi) Leak check of metering system
shown in figure 103 under R 336.2021. That portion of the sampling train
from the pump to the orifice meter shall must be leak checked
before to initial use and after each shipment. Leakage after the pump will
result in less volume being recorded than is actually sampled. The following
procedure is suggested,
also
(see figure 107 under R 336.2021): Close the main valve on
the meter box. Insert a 1-hole rubber stopper with rubber tubing attached into the
orifice exhaust pipe. Disconnect and vent the low side of the orifice
manometer. Close off the low side orifice tap. Pressurize the system to 13 to
18
centimeters, (5 to 7 in.ches,) water column by
blowing into the rubber tubing. Pinch off the tubing and observe the manometer
for 1 minute. A loss of pressure on the manometer indicates a leak in the meter
box.; leaks Leaks, if present, shall
must be corrected.
(vii) Barometer. Calibrate against a mercury barometer.
(f) Calculations. When carrying out
calculations, retain at least 1 extra decimal figure beyond that of the acquired
data. Round off figures after the final calculation. Other forms of the equations
may be used if they the other forms of the equations give equivalent
results. All
of the The
following
provisions under
R 336.2011(f) apply
to calculations: for this rule.
An = Cross-sectional area
of nozzle, m²(ft.²).
A = Cross-sectional area of stack or
flue at the point of sampling, ft².
B ws = Water vapor in the
gas stream, proportion by volume, expressed as a fraction.
B wi = Percent water vapor
in gas entering source particulate control device determined by method 4.
B wo = Percent water vapor
in gas exiting source particulate control device.
Ca = Wash blank residue
concentration, mg/g.
Cs = Concentration of
particulate matter in stack gas, pounds per 1,000 pounds of actual stack gas.
C sD = Concentration
of particulate matter in stack gas, moisture excluded, pounds per 1000
pounds of dry stack gas.
Cs50 = Concentration of
particulate matter corrected to 50% excess air, pounds per 1000 pounds of
stack gas.
Cs50D = Concentration of
particulate matter corrected to 50% excess air, excluding any water addition
from a collector, pounds per 1000 pounds of stack gas.
E = Mass emission rate of particulate,
lb/hr.
F50 = Concentration
conversion factor to 50% excess air with no moisture alterations in exhaust.
F50D = Concentration
conversion factor to 50% excess air, excluding any moisture added to exhaust
gas by pollution collection system.
FD = Concentration
conversion factor to dry basis, excluding any water in the stack gas.
I = Percent of isokinetic sampling.
L a = Maximum acceptable
leakage rate for either a pretest leak check or for a leak check following a
component change; equal to 0.00057 m³/min (0.02 cfm) or 4% of the average
sampling rate, whichever is less.
Li = Individual leakage
rate observed during the leak check conducted
before the "ith" component
change (i = 1, 2, 3 . . . . n), m³/min (cfm).
Lp = Leakage rate observed
during the post-test leak check, m³/min (cfm).
Md = Molecular weight of
dry stack gas, g/g mole (lb/lb-mole), calculated
by method 3, equation 3-21, using data from
integrated method 3.
mn = Total amount of
particulate matter collected, mg.
Mw = Molecular weight of
water, 18.0 g/g-mole (18.0 lb/lb-mole).
ma = Mass of residue of
solvent after evaporation, mg.
mg = Total weight of gas
samples through nozzle, lb.
P bar = Barometric pressure
at the sampling site, mm Hg (in. Hg).
Ps = Absolute stack gas
pressure.
Pstd = Standard absolute
pressure, 760 mm Hg (29.92 in. Hg).
R = Ideal gas constant, 0.06236
mm Hg-m³/°K-g-mole (21.85 in.Hg-ft.³/°R?lb-mole).
T m = Absolute average
dry-gas meter temperature (see figure 104), °K (°R).
Ts = Absolute average stack
gas temperature (see figure 104), °K (°R).
Tstd = Standard absolute
temperature, 294.I°K (530°R).
V a = Volume of solvent
blank, ml.
V aw = Volume of solvent
used in wash, ml.
V lc = Total volume of
liquid collected in impingers and silica gel (see figure 106), ml.
Vm = Volume of gas sample as
measured by the dry-gas meter, dcm (dcf).
V m(std) = Volume of gas
sample measured by the dry-gas meter, corrected to standard conditions, dscm
(dscf).
V w(std) = Volume of water
vapor in the gas sample, corrected to standard conditions, scm (scf).
V s = Stack gas velocity,
calculated by method 2, equation 2-9, using data obtained from method 5, m/sec
(ft./sec).
Wa = Weight of residue in
solvent wash, mg.
Y = Dry-gas meter calibration factor.
ΔH = Average pressure differential
across the orifice meter (see figure 104), mm H20 (in. H20).
%02 = Percent oxygen in
stack gas by volume (dry basis).
%N2 = Percent nitrogen in
stack gas by volume (dry basis).
p a = Density of solvent, mg/ml.
p s(std) = Density of all
sampled gas at standard conditions, lb/ft.³
pw = Density of water,
0.9982 g/ml (0.002201 lb/ml).
θ = Total sample time, min.
θ1 = Sample time, interval, from
the beginning of a run until the first component change, min.
θi = Sampling time interval,
between 2 successive component changes, beginning with the interval between
the first and second changes, min.
θp = Sampling time interval, from
the final (nth) component change until the end of the sampling run, min.
13.6 = Specific gravity of mercury.
60 = Sec/min.
100 = Conversion to percent.
386.9 = Cubic feet per lb-mole of
ideal gas at standard conditions.
453.6 = Conversion of pounds to grams.
3600 = Conversion of hours to sec.
1000 = Conversion of 1000 lb units to
lb units.
(ii) Average
the dry gas meter temperature and average the orifice pressure drop.
See data sheet (figure 104).
(iii) Dry gas
volume. Correct the sample volume measured by the dry gas meter to standard
conditions (21.1 degrees Centigrade, 760 mm Hg or 70
degrees Fahrenheit, 29.92 in. Hg) by using equation 5-1.
|
|
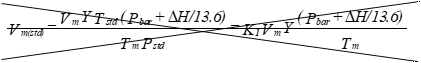
Where:
K1 =
0.3869 °K/mm Hg for metric units.
= 17.71
°R/in. Hg for English units.
Equation 5-1 can be used as
written. However, if the leakage rate observed during any of the mandatory
leak checks (for example, the post-test leak check or leak checks conducted
prior to component changes) exceeds La, equation 5-1 shall be
modified as follows:
(A) Case I. No component
changes made during sampling run. In this case, replace Vm in
equation 5-1 with the expression:
![]()
(B) Case II. One or more
component changes made during the sampling run. In this case, replace Vm in equation 5-1 by
the expression:
![]()
and substitute only for those leakage
rates (Li or Lp) that exceed La.
(iv) Volume of water
vapor.
|
|
Where:
K2 = 0.001338 m³/ml for metric
units.
= 0.04733 ft.³/ml for English units.
(v) Moisture content.
|
|
In saturated or water
droplet-laden gas streams, 2 calculations of the moisture content of the stack
gas shall be made: 1 from the impinger analysis (equation 5-3), and a second
from the assumption of saturated conditions. The lower of the 2 values of Bws
shall be considered correct. The procedure for determining the moisture
content based upon assumption of saturated conditions is given in the note of
section 1.2 of method 4. For the purpose of this method, the average stack gas
temperature from figure 104 may be used to make the determination, if
the accuracy of the in-stack temperature sensor is ±1 degree Centigrade (2
degrees Fahrenheit).
(vi) Solvent blank
concentration.
|
|
(vii) Solvent wash
blank.
|
|
(viii) Total
particulate weight. Determine the total particulate catch from the sum of the
weights obtained from containers 1, 1A, and 2 less the wash solvent blank (see
figure 106).
Refer to subdivision (d)(i)(E) of
this rule to assist in the calculation of results involving 2 or more pairs of
filters or 2 or more sampling trains.
(ix) Sampled gas
density. Determine the density of the gas sampled from the stack, at standard
conditions (lb/ft.³).
|
|
(x) Total weight of gas sampled
(lbs).
|
|
(xi) Particulate
concentration (lbs/1000 lbs).
|
|
(xii) Excess air and
moisture correction factors:
(A) Correction factor to 50%
excess air for those sources with or without any particulate collector where no
increase in moisture content of the exhaust gas occurs after the process and
before the point of sampling.
|
|
(B) Correction factor to 50%
excess air for those sources with a wet collection device (scrubber) that
increases the moisture content of the exhaust gas after the process and before
the point of sampling.
equation 5-10
|
|
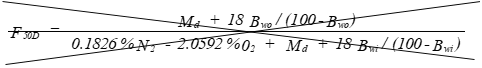
(C) Correction factor to convert
the actual concentration, Cs, to dry conditions.
equation 5-11
|
|
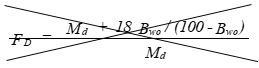
(xiii) Converted
particulate concentrations, where applicable under the department’s rules or
permit.
equation 5-12
|
|
(xiv) Mass emission
rate (lb/hr).
equation 5-15
|
|
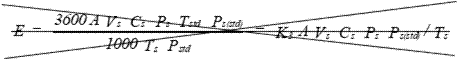
Where:
K3 = 63.77
for English units.
(xv) Isokinetic
variation:
(A) Calculation from raw
data.
equation 5-16
|
|
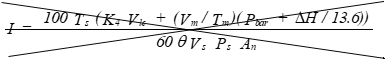
Where:
K4 = 0.003458 mm Hg - m³ ml - °K for metric
units.
= 0.002672
in. Hg - ft.³/ml - °R for English units.
(B) Calculation from
intermediate values.
equation 5-17
|
|
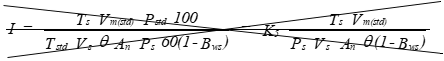
Where:
K5 = 4.307 for metric units.
= 0.09409 for English units.
(xvii) Acceptable results. If
90%=I=110%, the results are acceptable. If the results are low in comparison to
the standard and I is beyond the acceptable range, or if I is less than 90%, the
department may opt to accept the results. Otherwise, reject the results and
repeat the test.
(g) Bibliography:
(i) Federal Register, Volume 42, No. 160, Part 60, Chapter 1, Title 40, Appendix A Method 5, August 18, 1977.
(ii) Martin, Robert M. Construction Details of Isokinetic Source Sampling Equipment. Environmental Protection Agency. Research Triangle Park, N.C.APTD-0581. April, 1971.
(iii) Rom, Jerome J. Maintenance, Calibration, and Operation of Isokinetic Source Sampling Equipment. Environmental Protection Agency.Research Triangle Park, N.C. APTD-0576. March, 1972.
(iv) Shigehara, R. T. "Adjustments in the EPA Nomograph for Different Pitot Tube Coefficients and Dry Molecular Weights." Stack Sampling News 2:4-11.October, 1974.
(v) Guidelines for Source Testing of Particulate. Michigan Department of Natural Resources, Air Quality Division. June 1, 1977.
R336.2033 Test methods for coke oven quench towers.Rule 1033. (1) Test methods as applicable to coke oven quench towers. The publication entitled "Standard Methods for the Examination of Water and Wastewater,"(14th23rd edition),section 208C,shallmust apply to the measurement of total dissolved solids in coke oven quench tower water.
(2) In addition to the provisionsofin “Standard Methods for the Examination of Water and Wastewater” under subrule (1)section 208C, all of the following provisionsshallmust apply to the measurement of total dissolved solids in coke oven quench tower water:
(a) The quench tower makeup watershallmust be sampled at locations downstream of any makeup water additions.
(b) The quench tower watershallmust be sampled between the quench tower sump and the quench tower spray nozzles.
(c) One sample of quench tower water for all operating quench towersshallmust be collected once per day, 5 days per week.
(d) Compliance with the applicable quench tower limitsshallmust be determined on a weekly basis.
(e) For purposes of determining compliance, either individual analysis of the collected samples may be averaged or a weekly composite analysis may be performed. R 336.2040 Method for determination of volatile organic compound emissions from
coating lines and graphic arts lines.
Rule
1040. (1) The methods described in this rule shall must be used for the
determination of volatile organic compound emissions from coating lines and
graphic arts lines
for
the purpose of determining compliance, during the specified averaging period,
with an emission limit.
For emission limits expressed as pounds of volatile organic compounds per
gallon of coating, minus water, as applied. Concentrations of volatile organic
compounds in coatings and inks must be determined by excluding water and
compounds that are used as organic solvents and are excluded from the
definition of volatile organic compound from both the volume of volatiles in
the coatings and inks and the volume of the coatings and inks as applied. The emission
limits can be
contained in any of the following:
(a) These rules.
(b) A permit to install.
(c) A permit to operate.
(d) A voluntary agreement.
(e) A performance contract.
(f) A stipulation.
(g) An order of the department.
For emission limits expressed as
pounds of volatile organic compounds per gallon of coating, minus water, as
applied, the phrase "minus water" shall also include compounds which
are used as organic solvents and which are excluded from the definition of
volatile organic compound. Concentrations of volatile organic compounds in
coatings and inks shall be determined by excluding water and compounds which
are used as organic solvents and which are excluded from the definition of
volatile organic compound from both the volume of volatiles in the coatings and
inks and the volume of the coatings and inks as applied.
(2) Unless otherwise specified in these
rules or in a legally enforceable permit, order, or contract as described in
subrule (1) of this rule, for a particular coating line or graphic arts line,
the applicable method for the determination of volatile organic emissions from
coating lines and graphic arts lines is based upon on the form of the
specified emission limit as follows:
(a) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, use the method described in subrule (12)(a) of this rule.
(b) For coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, use the method described in subrule (12)(b) of this rule.
(c) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, use the method described in subrule (12)(c) of this rule.
(d) For coating lines with 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, use the method described in subrule (12)(d) of this rule.
(e) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, use the method described in subrule (12)(e) of this rule.
(f) For coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, use the method described in subrule (12)(f) of this rule.
(g) For graphic arts lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, as applied, use the method described in subrule (12)(g) of this rule.
(h) For graphic arts lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, as applied, use the method described in subrule (12)(h) of this rule.
(i) For flatwood paneling coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per 1,000 square feet of coated finished product, use the method described in subrule (12)(i) of this rule.
(j) For flatwood paneling coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per 1,000 square feet of coated finished product, use the method described in subrule (12)(j) of this rule.
(3) The following nomenclature applies to the equations described in this rule:
(a) a = An individual duct through which gases containing volatile organic compounds are ducted to an add-on emissions control device.
(b) B = Modified emission limit, converted from pounds of volatile organic compounds per gallon of coating, minus water, as applied, to pounds of volatile organic compounds per gallon of coating solids as applied.
(c) Cim
= Concentration of volatile organic compounds, as measured by the applicable
method, in the effluent gas flowing through stack "m" leaving the
add-on emissions control device, (parts per million by volume).
(d) Cza
= Concentration of volatile organic compounds, as measured by the applicable
method, in the influent gas flowing through duct "a" entering the
add-on emissions control device, (parts per million by volume).
(e) Dci =
Density of each ink or coating "i" as received from the ink or
coating supplier, (pounds
per gallon of ink or coating).
(f) Dsj
= Density of each volatile organic compound dilution solvent "j"
added to the coating, (pounds
per gallon of solvent, minus water).
(g) DE = Volatile organic compound destruction efficiency of the add-on emissions control device.
(h) E = Emission limit expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied.
(i) GT
= Total volume of all coatings "z" used during the averaging period, (gallons
of coating, minus water, as applied).
(j) i = An individual coating used during the averaging period that belongs to the coating category for which a compliance determination is being made pursuant to the provisions of this rule. For a graphic arts line, "i" is an individual ink or coating used during the averaging period.
(k) j = An individual dilution solvent used during the averaging period that is used in conjunction with a coating "i".
(l) k = An individual method of application of coating solids.
(m) Lci
= Volume of each coating "i" used during the averaging period, (gallons
of coating, minus water, as applied).
(n) Ldi
= Volume of each ink or coating "i" as received from the ink or
coating supplier and used during the averaging period, (gallons
of ink or coating).
(o) Lik
= Volume of each coating "i" used by each application method
"k" during the averaging period, (gallons of coating, minus water,
as applied).
(p) Lsj
= Volume of each volatile organic compound dilution solvent "j" added
to the coating during the averaging period, (gallons of solvent, minus
water).
(q) M = Total
weight of volatile organic compounds in all coatings "z" used during
the averaging period for a coating line or a graphic arts line, (pounds).
(r) m = An individual stack through which gases containing volatile organic compounds are ducted to the ambient air from an add-on emissions control device.
(s) Mr
= Total weight of volatile organic compounds recovered from a recovery-type
add-on emissions control device during the averaging period, (pounds).
(t) N =
Fraction, by weight, of the total volatile organic compounds emitted from an
operation which is captured and enters the add-on emissions control device, (pound per
pound).
(u) NTr
= Fraction, by weight, of the total volatile organic compounds in all coatings
"z" used during the averaging period for a coating line or a graphic
arts line which is controlled by an add-on emissions control device "r," (pound
per pound).
(v) P = For an individual coating "i," pounds of volatile organic compounds per gallon of coating, minus water, as applied.
(w) Ps = For an individual coating "i," pounds of volatile organic compounds, minus water, as received from the ink or coating supplier.
(x) Pa = As used in subrule (12)(a) of this rule for coating lines that do not have an add-on emissions control device, volume-weighted average pounds of volatile organic compounds per gallon of coating, minus water, as applied, for a single coating category during the averaging period.
(y) Pb = As used in subrule (12)(b) of this rule for coating lines that have 1 or more add-on emissions control devices, volume-weighted average pounds of volatile organic compounds per gallon of coating, minus water, as applied, for a single coating category during the averaging period.
(z) Pc = As used in subrule (12)(c) of this rule for coating lines that do not have an add-on emissions control device, volume-weighted average pounds of volatile organic compounds per gallon of coating solids, as applied, for a single coating category during the averaging period.
(aa) Pd = As used in subrule (12)(d) of this rule for coating lines that have 1 or more add-on emissions control devices, volume-weighted average pounds of volatile organic compounds per gallon of coating solids, as applied, for a single coating category during the averaging period.
(bb) Pe = As used in subrule (12)(e) of this rule for coating lines that do not have an add-on emissions control device, volume- weighted average pounds of volatile organic compounds per gallon of applied coating solids for a single coating category during the averaging period.
(cc) Pf = As used in subrule (12)(f) of this rule for coating lines that have 1 or more add-on emissions control devices, volume-weighted average pounds of volatile organic compounds per gallon of applied coating solids for a single coating category during the averaging period.
(dd) Pg = As used in subrule (12)(g) of this rule for graphic arts lines that do not have an add-on emissions control device, average pounds of volatile organic compounds per pound of solids, as applied, for all inks and coatings used during the averaging period.
(ee) Ph = As used in subrule (12)(h) of this rule for graphic arts lines that have 1 or more add-on emissions control devices, average pounds of volatile organic compounds per pound of solids, as applied, for all inks and coatings used during the averaging period.
(ff) Pi = As used in subrule (12)(i) of this rule for flatwood paneling coating lines that do not have an add-on emissions control devices, volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product for a single-coating category during the averaging period.
(gg) Pj = As used in subrule (12)(j) of this rule for flatwood paneling coating lines that have 1 or more add-on emissions control devices, volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product for a single-coating category during the averaging period.
(hh) Qim
= Volumetric flow rate of the effluent gas flowing through stack "m"
leaving the add-on emissions control device, (dry standard cubic feet
per hour).
(ii) Qza
= Volumetric flow rate of the influent gas flowing through duct "a"
entering the add-on emissions control device, (dry standard cubic feet
per hour).
(jj) Q3n
= Volumetric flow rate of the effluent gas leaving an uncontrolled stack
"n," (dry
standard cubic feet per hour).
(kk) r = An individual add-on emissions control device.
(ll) Rr = Reduction efficiency of a single add-on emissions control device.
(mm) Rt = Overall reduction efficiency of all add-on emissions control devices used for a coating line or a graphic arts line.
(nn) s =The total number of different add-on control devices "r" on a coating line or graphic arts line.
(oo) Ssq = The total surface
area of coated finished product for a single-coating category for a flatwood
paneling coating line during the averaging period, (square
feet).
(pp) T = Overall transfer efficiency for all coatings "i" for a single-coating category on a coating line for the averaging period.
(qq) t = The total number of stacks "m" leaving an add-on emissions control device "r".
(rr) Ti
= Transfer efficiency for application of coating "i," (%).
(ss) u = Total number of ducts "a" entering an add-on emissions control device "r".
(tt) Uci = For representative colors and parts that are tested for transfer efficiency, "Uci" is the volume of each representative color of coating that is applied to each representative part on a coating line during the averaging period.
(uu) V = For a
coating line, the volume of solids in all coatings used "zc" during
the averaging period, (gallons).
(vv) Vci
= Proportion of solids by volume in each coating "i," (gallon
of solids per gallon of coating, minus water, as applied).
(ww) W = For a
graphic arts line, the weight of solids in all inks and coatings used
"zg" during the averaging period, (pounds).
(xx) Wci
= Proportion of volatiles, (volatile organic compounds, water, and exempt
compounds), by weight in each
ink or coating "i" as received from the ink or coating supplier, (pound of
volatiles per pound of coating).
(yy) x = The total number of different application methods "k."
(zz) y = The total number of different dilution solvents "j."
(aaa) z = The total number of different coatings "i" used on a coating line or different number of inks and coatings "i" used on a graphic arts line during the averaging period. "z" is used generically in the equations specified in this rule for "zc" when the calculation is made for a coating line and for "zg" when the calculation is made for a graphic arts line.
(bbb) zc = The total number of different coatings "i" in the same coating category used during the averaging period.
(ccc) zg = The total number of different inks and coatings "i" used during the averaging period.
(4) The following provisions apply to the calculations for a coating line or graphic arts line made pursuant to the methods described in this rule:
(a) When carrying out calculations, carry not less than 5 significant digits in intermediate calculations. Round off figures after the final calculation, rounding off calculated emission numbers to not less than 2, but not more than 3, significant figures.
(b) The
calculations for a coating line shall must include all of
the coatings which are in the same coating category and which are used during
the averaging period as specified in the applicable emission limit.
(c) Except as
specified in R 336.1624(5)(d), the calculations for a graphic arts line shall
must include all of
the inks and coatings that are used during the averaging period as specified in
the applicable emission limit.
(5) The volatile organic compound content of an ink or
coating, minus water, as applied, "P," shall must be determined
using any of the following methods:
(a) The
volatile organic compound content of an ink or coating, minus water, as
applied, "P," shall must be determined according to all of
the following provisions:
(i) The
volatile organic compound content, minus water, as applied, shall must be determined as
follows:
(A)
For a coating used on a coating line or a coating used on a graphic arts line,
the volatile organic compound content, minus water, as applied, shall must be determined
using federal reference method 24 or federal reference method 24Aa, as applicable to
the coating, as described in R 336.2004, or an alternate method approved
by the department.
(B)
For an ink that is used on a graphic arts line, the volatile organic compound
content, minus water, as applied, shall must be determined using federal
reference method 24 or federal reference method 24Aa, as
applicable, as described in R 336.2004.
(ii) The ink
or coating sample shall must be taken at a point where the
sample will be representative of the ink or coating material as applied.
(iii) The
sample shall must be stored in an
enclosed container that is not less than 1 pint in volume.
(iv) By using
a procedure that is acceptable to the department, the amount of any compound in
the sample that is excluded from the definition of volatile organic compound
may be quantified and subtracted from the total amount of volatiles in the
sample as determined by federal reference method 24, federal reference method
24Aa, or an alternate
method that is approved by the department. In this case, the volume of any
excluded compound in the sample shall must also be
subtracted from the volume of the ink or coating sample.
(b) Upon
written approval by the department, the volatile organic compound content of an
ink or coating may be determined from formulation data, which includes batch
composition information from the ink or coating manufacturer and the amount of
volatile organic compound dilution solvent added to the ink or coating before
application. In this case, "P" shall must be calculated
using the following equation:
|
|
(c) If a
coating or ink is tested by a federal reference method 24 or 24Aa analysis or by an
alternate method approved by the department and the results are different than
calculated through formulation data review, then the test method results shall
must be used for
determining compliance with the emission limit.
(6) The
weight of volatile organic compounds that are used during the averaging period shall
must be calculated
using the following equation, where "z" is the total number of
coatings used:
|
|
(7) The
total volume of coating solids that are used during the averaging period shall
must be calculated
using the following equation, where "z" is the total number of
coatings used:
|
|
(8) The
total weight of ink and coating solids that are used during the averaging
period shall must be calculated
using the following equation, where "zg" is the total number of inks
and coatings used during the averaging period:
|
|
(9) The
transfer efficiency shall must be determined by the following
method, if approved by the department, or by an alternate method approved by
the department:
(a) A person who that is responsible
for the transfer efficiency test shall identify all of the following in a
coating operation transfer efficiency test proposal and shall submit the
proposal to the department for approval not less than 30 days before the
transfer efficiency test:
(i) All processing sequences. A processing sequence is the combination and order of paint booths, flash-off areas, ovens, and application equipment necessary to apply a coating.
(ii) The coating categories used on each processing sequence.
(iii) The
representative coating color in each coating category. A representative color shall
must be determined
based upon on the volume of
coating used in relation to the total volume of coating category coatings used
and any other parameters acceptable to the department. If it is believed by the
department that the transfer efficiency of the various coatings within the same
coating category would be different as a result of different coating
technologies, such as for metallic topcoat coatings and nonmetallic topcoat
coatings, 2 or more representative coating colors may be required by the
department to be tested.
(iv) At a
minimum, 2 different representative parts coated in each processing sequence. A
representative part shall must be determined based on the numbers
of the part coated in relation to the total number of parts coated, the
configuration of the part, and any other parameters acceptable to the
department.
(b) For the initial transfer efficiency tests, a person shall test, at a minimum, the representative color or colors in each coating category used on each representative part in each processing sequence. To more closely represent actual process conditions, coatings applied wet-on-wet, such as basecoat and clearcoat, may be tested together. Also, identical colors or clear coats on identical parts in identical processing sequences need not be tested.
(c) A person who that is responsible for
the transfer efficiency test shall review the operating conditions annually thereafter
after
the initial transfer efficiency tests and demonstrate, to the satisfaction of
the department, that significant changes have not occurred in coating
technology, the parts coated, or the processing sequence. The most recent test
results shall remain valid for 5 years if the person demonstrates that
significant change has not occurred. Significant product, processing, material,
or application equipment changes shall necessitate retesting of the
transfer efficiency of the operations that have been modified. The retesting shall
must be done as soon
as practicable, but not more than 180 days after the start-up and stabilization
of the new product, process, material, or application equipment. New transfer
efficiency values determined by the retest shall must be used
retroactively to the start-up of the new product, process, material, or
application equipment.
(d) Retests on
a coating line may be limited to a representative coating on not less than 2
representative parts in a representative processing sequence, as approved by
the department. A representative processing sequence shall must be determined
based upon on coating usage,
application equipment, and any other parameters acceptable to the department.
(e) The area in
the facility to be used for part weighing shall must be selected so as
to provide for an area that has relatively constant temperature and minimal air
movement.
(f) Except as
allowed pursuant to the provisions of subdivision (b) of this subrule, the
coating being tested shall must be the only coating applied during
the transfer efficiency test. If the part is weighed, weight loss from all
other materials shall must be accounted for in the initial
test weight.
(g) A minimum
of 10 parts per transfer efficiency test shall must be weighed to
determine the weight of the solids applied. The average weight gain for the 10
parts tested shall must be used as the part weight gain for that
transfer efficiency test. All transfer efficiency tests for a processing
sequence shall must be completed
within a 36-hour period.
(h) Each part
to be painted shall must be identified and preweighed to the
nearest 0.05 pound.
(i) The
selected paint material at each paint system or paint pot dedicated for the
transfer efficiency test shall must be isolated.
(j) The amount
of material used during the transfer efficiency test shall must be determined by
either of the following measurement procedures:
(i) The weight measurement procedure as follows:
(A) Weigh the tank of reduced paint, to the nearest 0.01 pound, after all supply and return lines have been filled.
(B) Connect the paint tank to the system and paint the test parts.
(C) Reweigh the tank to the nearest 0.01 pound.
(D) Determine the weight of paint material used by subtracting the final weight of the tank from the initial weight of the tank.
(E) Obtain paint samples for weight solids determination.
(F) Do both the initial and final weighings of the paint tank with the tank pressurized or with the tank not pressurized.
(ii) A volume measurement procedure that is acceptable to the department.
(k) Each
painted test part shall must be reweighed to the nearest 0.01
pound after paint has cured and cooled.
(l) The weight
of the solids in the paint samples shall must be determined
using ASTM‑D2369,
adopted by reference in R 336.1902. ASTM‑D2369 is adopted in
these rules by reference. A copy of this document may be inspected at the
Lansing office of the air quality division of the department of environmental
quality. A copy of this document may be obtained from the American
Society for Testing and Materials, 100 Barr Harbor Drive, West
Conshohocken, Pennsylvania 19428, or from the Department of Environmental
Quality, Air Quality Division, P.O. Box 30260, Lansing, Michigan 48909-7760, at
a cost as of the time of adoption of these rules of $25.00.
(m) The coating
density in pounds per gallon shall must be determined
using ASTM‑D1475,
adopted by reference in R 336.1902. ASTM‑D1475 is adopted in
these rules by reference. A copy of this document may be inspected at the Lansing
office of the air quality division of the department of environmental quality. A
copy of this document may be obtained from the American
Society for Testing and Materials, 100 Barr Harbor Drive, West
Conshohocken, Pennsylvania 19428, or from the Department of Environmental
Quality, Air Quality Division, P.O. Box 30260, Lansing, Michigan 48909-7760, at
a cost as of the time of adoption of these rules of $25.00.
(n) The
following equation shall must be used to calculate the transfer
efficiency for the application of coating "i":
|
|

Ti
=
![]()
(o) Where more
than 1 part type or coating are tested on a coating line for a single coating
category, the overall transfer efficiency "T" for the coating
category shall must be determined by
averaging the individual transfer efficiency values based upon on a volume-weighted
average of coatings applied during the averaging period for each different
color and part type tested. This overall transfer efficiency shall must be calculated by
using the following equation, where "aa" is the number of coatings
tested and "bb" is the number of part types tested:
|
|
(p) Baseline
operating parameters of the paint application equipment and the paint booths shall
must be established
for each transfer efficiency test and shall must serve as a basis
for determining compliance. These parameters shall must be included in a
transfer efficiency test report and shall must include all of
the following information:
(i) Type of spray equipment.
(ii) Electrostatic voltage.
(iii) Size and geometry of the part coated.
(iv) Gun-to-target
distance, (nonmanual).
(v) Number of parts per conveyor hook.
(vi) Air
velocity in spray booth, (linear feet per minute).
(vii) Fluid
flow settings, (by color).
(viii) Bell revolutions per minute for minibells.
(ix) Atomizing air pressure.
(10) The
capture efficiency "N" shall must be determined as
follows:
(a) The capture
efficiency "N" shall must be determined by using the methods
specified in 40 C.F.R.CFR §52.741(a)(4)(iii) entitled
"Capture system eEfficiency tTest pProtocols" and
in appendix B entitled "VOMolatile Organic Material Measurement
Techniques for Capture Efficiency," with the following modifications:
(i) The general modifications are as follows:
(A) Replace the requirements under 40 CFR 52.741(a)(4)(iii)(Aa) (2) with the following
requirements: If
a source owner or operator uses a control device designed to collect and
recover vocvolatile
organic compounds,
for example, carbon adsorber, an explicit measurement of capture efficiency is
not necessary if the conditions described in 40 C.F.R.CFR §52.741(a)(4)(iii)
are met. The overall emission reduction efficiency of the control system shall
must be determined
each day by directly comparing the input liquid vocvolatile organic
compounds
to the recovered liquid vocvolatile organic compounds. The procedure
for use in this situation is given in 40 C.F.R.CFR §60.433,
with the following modifications to 40 CFR
52.741(a)(4)(iii)(A)(2)(i) under paragraph (B) of this rule.:
(B)
Replace the requirements under 40 CFR 52.741(a)(4)(iii)(A)(2)(i) with the following
requirements:
The source owner or operator shall obtain data each day for the solvent usage
and solvent recovery and determine the solvent recovery efficiency of the
system each day using a 7-day rolling period. The recovery efficiency for each
day is computed as the ratio of the total recovered solvent for that day and
the prior 6 consecutive operating days to the total solvent usage for the same
7-day period weighted average as given in 40 C.F.R.CFR §60.433. This
ratio shall must be expressed as a
percentage. The ratio shall must be computed within 72 hours after
each 24-hour period. With the approval of the administrator, a source that
believes that the 7-day rolling period is not appropriate may use an
alternative multi-day rolling period of not more than 30 days.
(C)
Requirements
in 40 CFR 52.741
Aappendix B Ffor procedures G.1,
G.2, F.2, F.1, and L, the following modifications:sections titled “1.4 Sampling
requirements”. must be replaced
with the following:
A capture efficiency test shall must consist of not less than 3
sampling runs. Each run shall must cover at least 1 complete
production cycle, but shall must be not less than 3 hours long. The
sampling time for each run need not be more than 8 hours, even if the
production cycle has not been completed. Alternative sampling times may be used
if approved by the administrator.
For procedure L, the following
addition: 5.4 Audit procedure. Concurrently, analyze the audit sample
and a set of compliance samples in the same manner to evaluate the technique of
the analyst and the standards preparation. The same analyst, analytical
reagents, and analytical system shall be used both for compliance samples and
the EPA audit sample. If this condition is met, the auditing of subsequent
compliance analyses for the same enforcement agency within 30 days is not
required. An audit sample set shall not be used to validate different sets of
compliance samples under the jurisdiction of different enforcement agencies,
unless prior arrangements are made with both enforcement agencies.
For procedures G.1, G.2, F.2, F.1,
and L, the following additions:
5.5 (5.6 for procedure G.2) Audit
samples. Audit sample availability. Audit samples will be supplied only to
enforcement agencies for compliance tests. The availability of audit samples
may be obtained by writing to the following address:
Source Test Audit Coordinator
(MD-77B)
Quality Assurance Division
Atmospheric Research and Exposure
Assessment Laboratory
U.S. Environmental Protection
Agency
Research Triangle Park, NC 27711
The availability of audit samples
may also be obtained by calling the source test audit coordinator (STAC) at
(919) 541-7834. The request for the audit sample shall be made not less than 30
days before the scheduled compliance sample analysis.
5.6 (5.7 for procedure G.2) Audit
results. Calculate the audit sample concentration according to the calculation
procedure described in the audit instructions included with the audit sample. Fill
in the audit sample concentration and the analyst's name on the audit response
form included with the audit instructions. Send 1 copy to the EPA regional
office or the appropriate enforcement agency and a second copy to the STAC. The
EPA regional office or the appropriate enforcement agency will report the
results of the audit to the laboratory being audited. Include this response
with the results of the compliance samples in relevant reports to the EPA
regional office or the appropriate enforcement agency.
(ii) Owners or
operators of coating lines that have multiple stacks may choose to apply 1 of the 4 protocols under
40 CFR 52.741(a)(4)(iii)(B) or the protocol described under subparagraph (A) of
this paragraph, following
modifications in
addition to the modifications listed in paragraph (i) of this subdivision except for and to replace the modification
to 1.4 listed in paragraph (i)(C)(1) of this subdivision, which is
replaced with the following language as described under subparagraph (B) of
this paragraph:
(A)(a)(4)(iii)(B)
The capture efficiency of a coating line shall be measured using 1 of the 5
protocols given below. Any error margin associated with a test protocol shall
not be incorporated into the results of a capture efficiency test. If these
techniques are not suitable for a particular process, then the source shall
present an alternative capture efficiency protocol and obtain approval for it
by the administrator as a sip or fip revision.
(5) Liquid/gas method
measuring the captured emission, Gw, and liquid input, L. This
procedure may only be used when the capture efficiency for a coating line is
expected to be less than 50%. The capture efficiency equation to be used for
this protocol is:
CE = Gw/L
Where:
CE = Coating line capture efficiency, decimal fraction.
Gw = Mass of vom captured and delivered to a control device.
L = Mass of liquid vom input to coating line.
Procedure
G.1 contained in appendix B of this section is used to obtain Gw. Procedure
L contained in appendix B or the alternate method in R 336.2007 shall must be used to
determine L.
(B) Requirements in 40 CFR 52.741 Aappendix B fFor procedures G.1,
G.2, F.2, F.1, and L, under
the procedure section titled the following modification: “1.4 Sampling
requirements” must be replaced
with the following requirements:. A capture efficiency test shall must consist of not
less than 3 sampling runs. Each run shall must cover at least 1
complete production or processing cycle or shall must be at least 1
hour in duration. For automotive surface coating operations, the sampling time
per test shall must be based on
coating a minimum of 3 representative vehicles.
(b) The
test protocols and 40
CFR 52.741(a)(4)(iii) appendix B of 40 C.F.R. §52.741(a)(4)(iii) are adopted by reference under
R 336.1902.
in these rules by reference. A copy of these regulations may be inspected at
the Lansing office of the air quality division of the department of
environmental quality. A copy of these regulations may be obtained from the
Department of Environmental Quality, Air Quality Division, P.O. Box 30260,
Lansing, Michigan 48909-7760, at no charge. A copy of 40 C.F.R. part 52 may be
obtained from the Superintendent of Documents, Government
Printing Office, P.O. Box
371954, Pittsburgh, Pennsylvania 15250-7954, at a cost as of the time
of adoption of these rules of $36.00, or
on the United States government
printing office internet web site at http://www.access.gpo.gov.
(c) Where
multiple capture systems are used on a coating line or a graphic arts line, the
appropriate capture efficiency for each application method shall must be determined and
the overall capture efficiency for the coating line shall must be based upon
on
a
mass-weighted average of all volatile organic compounds used on the coating
line or the graphic arts line during the averaging period.
(11) The
overall reduction efficiency of add-on emissions control devices shall must be determined
using 1 or more of the following methods, as applicable:
(a) When a
destructive-type add-on emissions control device is used, the reduction
efficiency for the add-on emissions control device shall must be determined by
using the following method:
(i) The destruction efficiency of the add-on emissions control device is calculated by using the following equation, where "u" is the total number of ducts entering the control device and "t" is the total number of stacks leaving the control device:
|
|
(ii) Using the destruction efficiency as determined in paragraph (i) of this subdivision, the reduction efficiency of the add-on emissions control device is calculated by using the following equation:
|
|
(iii) If there is only 1 add-on emissions control device used on a coating line or graphic arts line, use the value calculated for "Rr" pursuant to paragraph (ii) of this subdivision as the value for RT in subsequent calculations.
(iv) The
concentration of volatile organic compound emissions entering and exiting the
add-on emissions control device shall must be determined by
using federal reference methods 25 or 25aA,
federal reference method 18 if approved by the department, the alternate version
of federal reference method 25 incorporating the Byron analysis, as described in
R 336.2004,
or an alternate method that is acceptable to the department. Ffederal
reference methods 1, 2, 3, and 4, as described in R
336.2004,
shall must be used as applicable for the
determination of the volumetric flow rate in the effluent gas. Alternate
federal reference method 1A, 2A, 2C, or 2D shall be used where appropriate.
(b) When an
add-on emissions control device is used that recovers volatile organic
compounds, the reduction efficiency of the device shall must be determined by
using 1 of the following methods:
(i) A mass balance of the products used and the products recovered, using the following equation:
|
|
(ii) An alternate method that is acceptable to the department.
(iii) If there is only 1 add-on emissions control device used on a coating line or a graphic arts line, use the value calculated for "Rr" pursuant to paragraph (i) or (ii) of this subdivision as the value for RT in subsequent calculations.
(c) If there is more than 1 add-on emissions control device used on a coating line or a graphic arts line, calculate the overall reduction efficiency by using the following equation:
|
|
(12) Compliance
with the specified emission limit shall must be determined
using 1 of the following methods, as applicable, based upon on the form of the
emission limit:
(a) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, use either of the following methods:
(i) If only 1 coating is used on the coating line during the averaging time, use the following method:
(A) Determine the volatile organic compound content of the coating, minus water, as applied, "P," by using the method described in subrule (5) of this rule.
(B) If "P" is less than or equal to the specified emission limit, the coating line meets the emission limit.
(ii) If more than 1 coating of the same coating category is used on the coating line during the averaging period, use the following method:
(A) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(B) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(C) Determine the total volume of coatings used on the coating line during the averaging period "GT" using the following equation:
|
|
(D) Determine the volume-weighted average weight of volatile organic compounds per gallon, minus water, as applied, by using the following equation:
|
|
(E) If "Pa" is less than or equal to the specified emission limit, the coating line meets the emission limit.
(b) For coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, use the following method:
(i) Convert the specified emission limit to a modified emission limit "B" expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, by using the following equation:
|
|
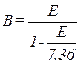
(ii) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(iii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iv) Determine the total volume of coating solids used during the averaging period "V" by using the method described in subrule (7) of this rule.
(v) Determine the overall capture efficiency "N" by using the method described in subrule (10) of this rule.
(vi) Determine the overall reduction efficiency "RT" by using the method described in subrule (11) of this rule.
(vii) Determine the volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pb," by using the following equation:
|
|
(viii) If "Pb" is less than or equal to the modified limit "B," the coating line meets the emission limit.
(c) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total volume of coating solids used during the averaging period "V" by using the method described in subrule (7) of this rule.
(iv) Determine the volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pc," by using the following equation:
|
|
(v) If "Pc" is less than or equal to the specified limit, the coating line meets the emission limit.
(d) For coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total volume of coating solids used during the averaging period "V" by using the method described in subrule (7) of this rule.
(iv) Determine the overall capture efficiency "N" by using the method described in subrule (10) of this rule.
(v) Determine the overall reduction efficiency "RT" by using the method described in subrule (11) of this rule.
(vi) Determine the volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pd," by using the following equation:
|
|
(vii) If "Pd" is less than or equal to the specified limit, the coating line meets the emission limit.
(e) For coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total volume of coating solids used during the averaging period "V" by using the method described in subrule (7) of this rule.
(iv) Determine the overall transfer efficiency "T" by using the method described in subrule (9) of this rule.
(v) Determine the volume-weighted average weight of volatile organic compounds per gallon of applied coating solids "Pe" by using the following equation:
|
|
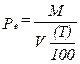
(vi) If "Pe" is less than or equal to the specified limit, the coating line meets the emission limit.
(f) For coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total volume of coating solids used during the averaging period "V" by using the method described in subrule (7) of this rule.
(iv) Determine the overall transfer efficiency "T" by using the method described in subrule (9) of this rule.
(v) Determine the overall capture efficiency "N" by using the method described in subrule (10) of this rule.
(vi) Determine the overall reduction efficiency "RT" by using the method described in subrule (11) of this rule.
(vii) Determine the volume-weighted average weight of volatile organic compounds per gallon of applied coating solids "Pf" by using the following equation:
|
|
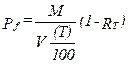
(viii) If "Pf" is less than or equal to the specified limit, the coating line meets the emission limit.
(g) For graphic arts lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, as applied, use the following method:
(i) Determine the volatile organic compound content of each ink and coating, minus water, as applied, "P," used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the weight of ink and coating solids used during the averaging period "W" by using the method described in subrule (8) of this rule.
(iv) Determine the average pounds of volatile organic compound per pound of solids, as applied, "Pg," by using the following equation:
|
|
(v) If "Pg" is less than or equal to the specified limit, the graphic arts line meets the emission limit.
(h) For graphic arts lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, as applied, use the following method:
(i) Determine the volatile organic compound content of each ink and coating, minus water, as applied, "P," used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the weight of ink and coating solids used during the averaging period "W" by using the method described in subrule (8) of this rule.
(iv) Determine the overall capture efficiency "N" by using the method described in subrule (10) of this rule.
(v) Determine the overall reduction efficiency "Rt" by using the method described in subrule (11) of this rule.
(vi) Determine the average pounds of volatile organic compound per pound of solids, as applied, "Ph," by using the following equation:
|
|
(vii) If "Ph" is less than or equal to the specified limit, the graphic arts line meets the emission limit.
(i) For flatwood paneling coating lines that do not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compound per 1,000 square feet of coated finished product, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total surface area of coated finished product for the coating category during the averaging period "sq".
(iv) Determine the volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product "Pi" by using the following equation:
|
|
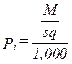
Where the units for the term 1,000 are square feet.
(v) If "Pi" is less than or equal to the specified limit, the coating line meets the emission limit.
(j) For flatwood paneling coating lines that have 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per 1,000 square feet of coated finished product, use the following method:
(i) Determine the volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period by using the method described in subrule (5) of this rule.
(ii) Determine the weight of volatile organic compounds used during the specified averaging period "M" by using the method described in subrule (6) of this rule.
(iii) Determine the total surface area of coated finished product for the coating category during the averaging period "sq".
(iv) Determine the overall capture efficiency "N" by using the method described in subrule (10) of this rule.
(v) Determine the overall reduction efficiency "RT" by using the method described in subrule (11) of this rule.
(vi) Determine the volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product "Pj" by using the following equation:
|
|
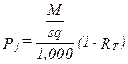
Where the units for the term 1,000 are square feet.
(vii) If "Pj" is less than or equal to the specified limit, the coating line meets the emission limit.
R 336.2041 Recordkeeping requirements for coating lines and graphic arts lines.Rule 1041. (1) Unless otherwise specified in any of the following, the recordkeeping requirements specified in this ruleshallmust apply to coating lines and graphic arts lines subject to emission limits contained in any of the following:
(a) These rules. (b) A permit to install. (c) A permit to operate. (d) A voluntary agreement. (e) A performance contract. (f) A stipulation. (g) An order of the department. (h) A renewable operating permit.(2) If a coating line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, and if only 1 coating is used on the coating line during the averaging time, then a person shall keep records of the name, identification number, volume "Lci," and volatile organic compound content of the coating, minus water, as applied, "P," used during the averaging period, as described in R 336.204-0(5). If this single coating used during an averaging period is in compliance with all of the emission limits specified in subrule (1) of this rule, then the volume "Lci" for the averaging period may be calculated, baseduponon coating usage records during a time period of not more than 1 month, with the coating usage prorated to the specified averaging period using a method approved by the department for that coating line.
(3) If a coating line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, and if more than 1 coating of the same coating category is used on the coating line during the averaging period, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period, as described in R 336.2040(5). If all coatings used on the coating line during an averaging period are in compliance with all of the emission limits specified in subrule (1) of this rule, then the volume "Lci" for the averaging period may be calculated, based upon coating usage records during a time period of not more than 1 month, with the coating usage prorated to the specified averaging period using a method approved by the department for that coating line. (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coatings used on the coating line during the averaging period "GT," as described in R 336.2040(12). (d) The volume-weighted average weight of volatile organic compounds per gallon, minus water, as applied, "Pa," as described in R 336.2040(12). (4) If a coating line has 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating, minus water, as applied, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coating solids and volume of ink or coating "Ldi" used during the averaging period "V," as described in R 336.2040(7). (d) The overall capture efficiency "N," as described in R 336.2040(10). (e) The overall reduction efficiency "Rt, " as described in R 336.2040(11), including the parameters "Qza," "Cza," "Qim," "Cim," and "Mr." (f) The volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pb," as described in R 336.2040(12). (5) If a coating line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coating solids and volume of ink or coating "Ldi" used during the averaging period "V," as described in R 336.2040(7). (d) The volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pc," as described in R 336.2040(12). (6) If a coating line has 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of coating solids, as applied, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coating solids and volume of ink or coating "Ldi" used during the averaging period "V," as described in R 336.2040(7). (d) The overall capture efficiency "N," as described in R 336.2040(10). (e) The overall reduction efficiency "Rt," as described in R 336.2040(11), including the parameters "Qza," "Cza," "Qim," "Cim," and "Mr." (f) The volume-weighted average weight of volatile organic compounds per gallon of coating solids, as applied, "Pd," as described in R 336.2040(12). (7) If a coating line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, then a person shall keep all of the following records:(a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "Pp" used during the averaging period, as described in R 336.2040(5).
(b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coating solids and volume of ink or coating "Ldi" used during the averaging period "V," as described in R 336.2040(7). (d) The overall transfer efficiency "T," as described in R 336.2040(9), including "Ti" and "Uci". (e) The volume-weighted average weight of volatile organic compounds per gallon of applied coating solids "Pe," as described in R 336.2040(12). (8) If a coating line has 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per gallon of applied coating solids, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, that belongs to the same coating category "P" used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total volume of coating solids and volume of ink or coating "Ldi" used during the averaging period "V," as described in R 336.2040(7). (d) The overall transfer efficiency "T," as described in R 336.2040(9), including "Ti" and "Uci". (e) The overall capture efficiency "N," as described in R 336.2040(10). (f) The overall reduction efficiency "Rt," as described in R 336.2040(11), including the parameters "Qza," "Cza," "Qim," "Vim," and "Mr." (g) The volume-weighted average weight of volatile organic compounds per gallon of applied coating solids "Pf," as described in R 336.2040(12). (9) If a graphic arts line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each ink and coating, minus water, as applied, "P," used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The weight of ink and coating solids used during the averaging period "W," as described in R 336.2040(8), including "Wci" and "Ldi." (d) The average pounds of volatile organic compound per pound of solids, as applied, "Pg," as described in R 336.2040(12). (10) If a graphic arts line has 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per pound of solids, as applied, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each ink and coating, minus water, as applied, "P," used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The weight of ink and coating solids used during the averaging period "W," as described in R 336.2040(8), including "Wci" and "Ldi." (d) The overall capture efficiency "N," as described in R 336.2040(10). (e) The overall reduction efficiency "Rt," as described in R 336.2040(11), including the parameters "Qza," "Cza," "Qim," "Cim," and "Mr." (f) The average pounds of volatile organic compound per pound of solids, as applied, "Ph," as described in R 336.2040(12). (11) If a flatwood paneling coating line does not have an add-on emissions control device for which emission limits are expressed in pounds of volatile organic compound per 1,000 square feet of coated finished product, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, "P," used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total surface area of coated finished product for the coating category during the averaging period "sq," as described in R 336.2040(3). (d) The volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product "Pi," as described in R 336.2040(12). (12) If a flatwood paneling coating line has 1 or more add-on emissions control devices for which emission limits are expressed in pounds of volatile organic compounds per 1,000 square feet of coated finished product, then a person shall keep all of the following records: (a) The name, identification number, volume "Lci," and volatile organic compound content of each coating, minus water, as applied, "P," used during the averaging period, as described in R 336.2040(5). (b) The weight of volatile organic compounds used during the averaging period "M," as described in R 336.2040(6). (c) The total surface area of coated finished product for the coating category during the averaging period "sq," as described in R 336.2040(3). (d) The overall capture efficiency "N," as described in R 336.2040(10). (e) The overall reduction efficiency "Rt," as described in R 336.2040(11), including the parameters "Qza," "Cza," "Qim," "Cim," and "Mr." (f) The volume-weighted average pounds of volatile organic compounds per 1,000 square feet of coated finished product "Pj," as described in R 336.20-40(12).(13) An owner or operator of primer surfacer or topcoat operations subject to emission limits in R 336.1610(11), table 62,or R 336.1610a(4), table 64-a, shall keep records as required in the publication entitled "Protocol for Determining the Daily Volatile Organic Compound Emission Rate of Automobile and Light-duty Truck Topcoat Operations," EPA-453/R-08/0020/3-88-018, December, 1988, which is referenced in R 336.1610(6)(b)adopted by reference in R 336.1902.
(14) The records that are required in this ruleshallmust be retained for a period of not less than 2 complete yearsfromafter the date of collection and,uponon request by the department,shallmust be submitted to the department in an acceptable format.